诺和力说明书
Liraglutide Injection
利拉鲁肽
化学名称:Arg34Lys26-(N-ε-(γ-Glu(N-α-十六酰基)))-GLP-1[7-37]
化学结构式:
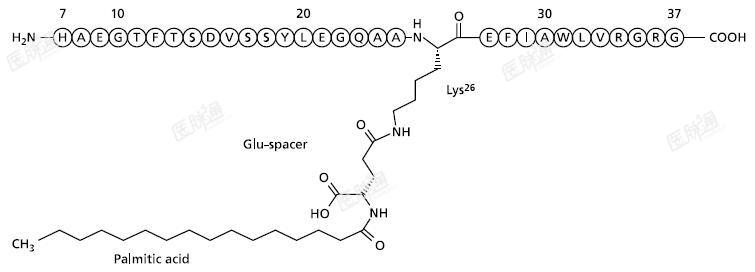
分子式:C172H265N43O51
分子量:3751.20Da
本品以苯酚作为抑菌剂,每 100 mL 本品中加入苯酚 0.55 g。
其他成份:二水合磷酸氢二钠、丙二醇、盐酸和或氢氧化钠(仅作为 pH 调节剂)和注射用水。
本品为无色或几乎无色的澄明等渗液体;pH=8.15
本品适用于成人2 型糖尿病患者控制血糖:
适用于单用二甲双胍或磺脲类药物最大可耐受剂量治疗后血糖仍控制不佳的患者,与二甲双胍或磺脲类药物联合应用。
适用于降低伴有心血管疾病的 2 型糖尿病成人患者的主要心血管不良事件(心血管死亡、非致死性心肌梗死或非致死性卒中)风险。
用量
为了改善胃肠道耐受性,利拉鲁肽的起始剂量为每天 0.6 mg。至少 1 周后,剂量应增加至 1.2 mg。预计一些患者在将剂量从 1.2 mg 增加至 1.8 mg 时可以获益,根据临床应答情况,为了进一步改善降糖效果,在至少一周后可将剂量增加至 1.8 mg。推荐每日剂量不超过 1.8 mg。
本品可用于与二甲双胍联合治疗,而无需改变二甲双胍的剂量。
本品可用于与磺脲类药物联合治疗。当本品与磺脲类药物联用时,应当考虑减少磺脲类药物的剂量以降低低血糖的风险。
调整本品的剂量时,无需进行自我血糖监测。然而,当本品与磺脲类药物联合治疗而调整磺脲类药物的剂量时,可能需要进行自我血糖监测。
特殊人群
肾功能受损患者:轻度、中度或重度肾功能受损的患者不需要进行剂量调整。在终末期肾病患者中无治疗经验,目前不推荐本品用于此类患者。
肝功能受损患者:轻度或中度肝功能受损患者不需要进行剂量调整。目前不推荐本品用于重度肝功能受损患者。
用法
本品每日注射一次,可在任意时间注射,无需根据进餐时间给药。本品经皮下注射给药,注射部位可选择腹部、大腿或者上臂。在改变注射部位和时间时无需进行剂量调整。然而,推荐本品于每天同一时间注射,应该选择每天最为方便的时间。更多有关给药的指导参见使用及其他操作的注意事项。
本品不可静脉或肌内注射。
附件:
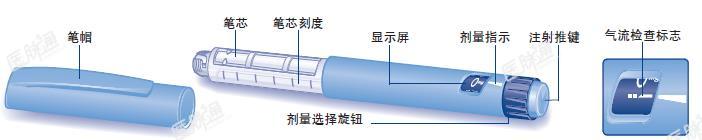
利拉鲁肽注射笔
操作指南
使用本品前请仔细阅读下面的操作指南
本品含 18 mg 利拉鲁肽。可供选择的剂量为 0.6 mg、1.2 mg 和 1.8 mg。
本品应与长度不超过 8 mm 以及细至 32 G(0.25/0.23 mm)的一次性诺和针®配合使用。
针头(示例)

维护本品
不要试图修理或拆开本品。
避免本品接触粉尘、污物或各种液体。
使用经温性洗涤剂浸湿的湿布擦拭本品。
请不要冲洗,浸洗或润滑本品,否则可能使本品受损。
重要信息
请勿与他人共同使用本注射笔或针头。
请将本品置于其他人,尤其是儿童不可触及处。
准备注射
检查注射笔的名称和彩色标签,确认其中含有利拉鲁肽。用药错误可能导致严重伤害。
A 摘下笔帽。

B 取一个新的一次性针头。揭掉针头上的保护片,直接将针头与本品拧紧。
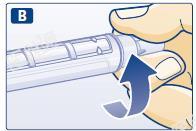
C 取下外针帽并保存,以便后续使用。
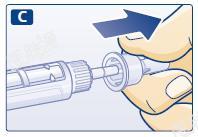
D 取下内针帽,并丢弃。
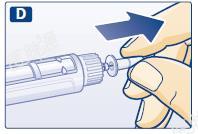
每次注射时请使用新的针头,从而避免污染、感染、漏液、针头堵塞和剂量不准确的风险。
在使用前请注意不要弄弯或损坏针头。
不要试图把已经取下的内针帽重新盖回针头上。您可能会刺伤自己。
排除空气
每次使用新注射笔进行注射前,应排除空气。如果您的注射笔已经使用过,请直接进行第 H 步“选择剂量”。
E 调节剂量选择旋钮,直至气流检查标志对准指针。

F 握住本品并使针尖向上,用手指轻弹笔杆数下,以使气泡都向上聚集至笔芯顶部。
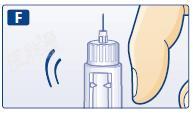
G 保持针尖向上,充分推压注射推键,使剂量刻度回复至 0。
针尖应出现一滴利拉鲁肽。如果无液滴出现,重复第 E 步至第 G 步操作,但不能超过 4 次。
如果仍未出现利拉鲁肽液滴,请更换针头,并重复第 E 步至第 G 步 1 次。
如果仍未出现利拉鲁肽液滴,请勿使用该注射笔。这表明注射笔损坏,必须更换新注射笔。
如果您的注射笔掉落至硬物表面或怀疑出现了某些问题,那么应更换一个新的一次性注射针头并在注射前排除空气。
选择剂量
检查:剂量选择旋钮应指示在零刻度。
H 调节剂量选择旋钮,使剂量指示在需注射的剂量(0.6 mg、1.2 mg 或 1.8 mg)。如果不慎选择了一个错误的剂量,只需通过向后或向前转动剂量选择旋钮直至剂量指示在正确剂量。回调旋钮时,注意不要触动注射推键,以免药液溢出。如果剂量选择旋钮在达到所需的剂量之前停止,说明所剩余的药液不足以进行足量的注射。此时,您可以:
将给药量分成两次进行注射:
将剂量选择旋钮向任一方向旋转,直至剂量指示在 0.6 mg 或 1.2 mg。注射该剂量。然后准备一支新的注射笔注射剩余的剂量数以完成足量注射。
只有在得到医疗专业人士的培训或建议后,方可将给药量通过现用注射笔和新注射笔分别进行注射。用计算器分配剂量。如果剂量分配错误,则利拉鲁肽的注射量会过多或过少。
使用新的注射笔进行足量注射:
如果剂量选择旋钮在指示在 0.6 mg 之前停止,那么需准备一支新注射笔注射足量药物。
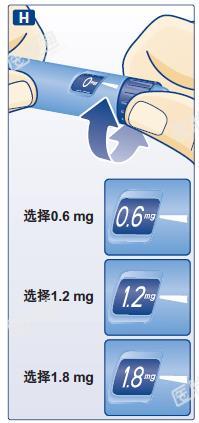
请勿选择 0.6 mg、1.2 mg 或 1.8 mg 之外的剂量。显示窗的数字必须准确对准剂量指示指针以确保您注射了正确的剂量。旋转剂量选择旋钮时会发出咔嗒的声音。不得使用这些咔嗒声来选择剂量。请勿使用笔芯刻度来计算药液的注射剂量,这样不够准确。
注射
将针头插入皮下。请遵照医生或护士指导的注射方法操作。然后遵照下列步骤进行:
I 注射时应将注射推键完全按下,直到剂量指示到 0 刻度。注意在注射期间勿使您的其他手指触摸到显示屏或在侧方按下剂量选择旋钮。这些动作会阻止注射。注射时,按住注射推键使针头在皮下停留至少 6 秒,以确保药液全部注射进入体内。

J 拔出针头。随后您可能看到针尖上有一滴药液。此为正常现象,不会影响您所注射的药物剂量。
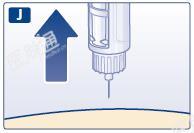
K 在不触碰针头或外针帽的情况下将针头放入外针帽。
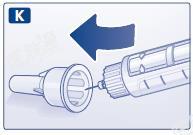
L 当针头全部进入外针帽后,小心将外针帽完全盖上,然后拧下针头,妥善弃之。套上笔帽。用过的本品,应在卸下针头之后再小心丢弃。请按照当地法规要求处置注射笔和针头。
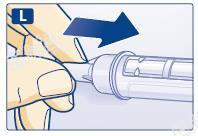
每次注射后和存放本品时应卸下针头。
这样可以降低本品污染、感染、漏液、针头堵塞和剂量不准确的风险。
处理使用过的针头时须非常小心以免刺伤和交叉感染。
安全性情况摘要
在 5 项大规模的长期 3a 期临床试验中,已有超过 2500 例患者接受了本品单药治疗或本品与二甲双胍、磺脲类药物(加或不加二甲双胍)或二甲双胍加罗格列酮联合治疗。 临床试验期间最常见的不良反应为胃肠道不适:恶心和腹泻十分常见,呕吐、便秘、腹痛和消化不良常见。在开始本品治疗时,这些胃肠道不良反应的发生频率可能更高。上述不良反应通常在治疗持续数天或数周内减轻。头痛和鼻咽类也是常见不良反应。此外,低血糖为常见不良反应,而当本品与磺脲类药物联用时则十分常见。重度低血糖主要发生在本品与磺脲类药物联用时。
不良反应列表
表 1 列出了本品长期 3a 期对照临床试验、LEADER 试验(一项长期心血管结局试验)和自发(上市后)报告的不良反应。所有事件的发生率均是基于 3a 期临床试验中的发生率进行计算的。
不良反应发生的频率定义如下:十分常见( ≥ 1/10);常见( ≥ 1/100,<1/10);偶见( ≥ 1/1000,<1/100),罕见( ≥ 1/10000,<1/1000);十分罕见(1/10000);不详(根据现有的数据无法评价)。在每个频率分组中,不良反应都是按照严重性降低的顺序列出。
表 1. 长期对照 3a 期、心血管结局试验(LEADER)中和自发(上市后)报告的不良反应
| MedDRA 22.0 | 十分常见 | 常见 | 偶见 | 罕见 | 十分罕见 |
| 感染与侵染 | 鼻咽炎 | ||||
| 免疫系统疾病 | 速发过敏反应 | ||||
| 代谢与营养疾病 | 低血糖 | 脱水 | |||
| 神经系统疾病 | 头痛 | ||||
| 心脏器官疾病 | 心率升高 | ||||
| 胃肠系统疾病 | 恶心 | 呕吐 | 肠梗阻 | 胰腺炎(包括坏死性胰腺炎) | |
| 肝胆系统疾病 | 胆石症 | ||||
| 皮肤及皮下组织类疾病 | 皮疹 | 荨麻疹 | |||
| 肾与泌尿系统疾病 | 肾损害 | ||||
| 全身性疾病及给药部位各种反应 | 疲乏 | 不适 | |||
| 各类检查 | 脂肪酶升高 |
来自对照 3b 和 4 期临床试验,仅来自对其测量的试验。
部分不良反应的描述
在一项本品单药治疗的临床试验中,本品治疗组低血糖的发生率低于活性对照组(格列美脲)。最常见的不良反应为胃肠道疾病以及感染及侵染类疾病。
低血糖
临床试验中大部分确认的低血糖事件均为轻度。未在本品单药治疗的试验中观察到重度低血糖事件。重度低血糖比较罕见,主要发生在本品与磺脲类药物联用时(0.02 事件患者年)。本品与磺脲类药物之外的口服抗糖尿病药物联用时所观察到的低血糖事件非常少(0.001 事件患者年)。在 LEADER 试验中,利拉鲁肽组报告的严重低血糖发生率低于安慰剂组(1.0 与 1.5 事件/100 患者年;率比估计值为 0.69[0.51-0.93])。
胃肠道不良反应
当本品与二甲双胍联用时,20.7% 的患者至少报告了 1 次恶心事件,12.6% 的患者至少报告了 1 次腹泻事件。当本品与磺脲类药物联用时,9.1% 的患者至少报告了 1 次恶心事件,7.9% 的患者至少报告了 1 次腹泻事件。大部分事件均为轻至中度,且呈剂量依赖性。大部分最初出现恶心症状的患者在继续治疗时,这些症状的频率和严重程度均有所降低。
在本品单药治疗的临床试验中,本品两个剂量组中的患者在开始治疗的前几周报告的恶心的发生率(14%)比格列美脲组患者恶心的发生率(3%)高。 接受本品治疗的患者恶心的发生率随时间的推移而降低,治疗 16 周后,利拉鲁肽组和格列美脲组患者恶心的发生率相似。 70 岁以上患者接受本品治疗时,可能会出现更多的胃肠道反应。
轻度和中度肾功能受损(肌酐清除率分别为 60-90 mL/min 和 30-59 mL/min)的患者接受本品治疗时,可能会出现更多的胃肠道反应。
胆石症和胆囊炎
在利拉鲁肽的长期、对照、3a 期临床试验中,报告的胆石症(0.4%)和胆囊炎(0.1%)病例极少。在 LEADER 试验中,利拉鲁肽组胆石症和胆囊炎的发生率分别为 1.5% 和 1.1%,安慰剂组分别为 1.1% 和 0.7%。
退出
在长期(26 周或更长)对照试验中,本品治疗组患者中由于不良反应导致的退出率为 7.8%,而在对照组患者中为 3.4%。本品治疗组中最常见的导致退出的不良反应为恶心(2.8%)和呕吐(1.5%)。
注射部位反应
在长期(26 周或更长)对照试验中,约 2% 接受本品的受试者报告了注射部位反应。这些反应通常都为轻度。
免疫原性
与其他含蛋白质或肽类的药物可能具有免疫原性相一致,患者在接受本品治疗之后可能会产生抗利拉鲁肽抗体。平均有 8.6% 的患者会产生抗体。抗体形成不会导致本品疗效的降低。
胰腺炎
在本品长期、对照、3 期临床试验期间已经报告了少数(<0.2%)急性胰腺炎病例。在本品上市后使用中也报告了胰腺炎病例。在 LEADER 试验中,经判定的急性胰腺炎发生率在利拉鲁肽组为 0.4%,安慰剂组为 0.5%。
过敏反应
在本品上市后使用中,已经报告了包括荨麻疹、皮疹和瘙痒在内的过敏反应。在本品上市后使用中,已经报告了少数伴随其他症状(如低血压、心悸、呼吸困难和水肿)的过敏反应。在本品所有长期临床试验中报告了少数(0.05%)血管性水肿。
报告疑似不良反应
药品批准上市后报告疑似不良反应十分重要,以便对药品的受益风险平衡进行持续监测。医疗专业人士应通过国家相关报告系统报告任何疑似不良反应。
以下患者禁用:
对本品活性成份或者本品中任何其他辅料过敏者。
警告:甲状腺C细胞肿瘤风险 • 利拉鲁肽在临床相关的暴露水平可能导致啮齿类动物甲状腺C细胞肿瘤。尚不清楚利拉鲁肽能否导致人类甲状腺C细胞肿瘤,包括甲状腺髓样癌(MTC),因为临床或非临床研究均无法确定其与人类的相关性。 • 利拉鲁肽不得用于有MTC既往史或家族史患者以及2型多发性内分泌肿瘤综合征患者(MEN2)。
本品不得用于 1 型糖尿病患者或用于治疗糖尿病酮症酸中毒。
本品并非胰岛素替代物。
本品不得用于有甲状腺髓样癌(MTC)既往史或家族史患者以及 2 型多发性内分泌肿瘤综合征患者(MEN2)。
尚无本品在纽约心脏病学会(NYHA)心功能分级为 IV 级的充血性心力衰竭患者中的治疗经验,目前不推荐本品用于此类患者。
在炎症性肠病和糖尿病性胃轻瘫患者中的治疗经验有限。不推荐本品用于这些患者,因为本品治疗过程中会伴随有一过性的胃肠道不良反应,包括恶心、呕吐和腹泻。
急性胰腺炎
使用 GLP-1 受体激动剂的患者观察到急性胰腺炎的发生。应当告知患者急性胰腺炎的特征性症状。如果怀疑发生了胰腺炎,应该停用本品;如果确认患者发生了急性胰腺炎,不应再使用本品进行治疗。
甲状腺疾病
临床试验中,尤其是既往有甲状腺疾病的患者中已经报告了甲状腺不良事件,例如甲状腺肿,此类患者应慎用本品。
低血糖
接受本品联合磺脲类药物治疗的患者发生低血糖的风险可能增加。减少磺脲类药物的剂量可以降低低血糖的风险。
脱水
接受本品治疗的患者已经报告了包括肾功能受损和急性肾衰竭在内的脱水的体征和症状。接受本品治疗的患者,应告知其治疗期间有发生胃肠道不良反应相关性脱水的潜在风险,应采取预防措施以避免体液耗竭。
对驾驶和机械操作能力的影响
本品对驾驶和机械操作能力没有或只有极小影响。应告知患者在驾驶和操作机械时预防低血糖发生,特别是当本品与磺脲类药物合用时。
使用和其他操作的特别注意事项
本品仅在呈无色或几乎无色澄明时才可使用。
本品不得在冷冻后使用。
本品应使用长度不超过 8 mm 以及细至 32 G 的针头给药。
本注射笔应与一次性的诺和针<sup>®</sup>配合使用。
本品不包含针头。
在临床试验和上市后使用中,已报告的药物过量所使用的剂量高达推荐维持剂量的 40 倍(72 mg)。总体上,患者报告了重度恶心、呕吐和腹泻。无重度低血糖的报告。所有患者均恢复并且没有出现并发症。
如果发生药物过量,应当根据患者的临床体征和症状采取适当的支持治疗。
尚未在18岁以下儿童和青少年中确定本品的安全性和有效性。尚未获得相关数据。根据一项在健康受试者中进行的药代动力学研究以及对患者(18 至 80 岁)的群体药代动力学数据分析的结果,年龄不会对利拉鲁肽的药代动力学产生与临床相关的影响。因此,不需要根据年龄进行剂量调整。目前在妊娠妇女中使用本品的数据尚不充分。动物试验结果提示本品具有生殖毒性。本品对人类的潜在风险尚不清楚。 本品不得在妊娠期间使用,此时推荐使用胰岛素。如果患者在治疗期间计划怀孕或已经怀孕,应停止本品治疗。利拉鲁肽是否在人乳中分泌,尚不清楚。动物试验结果提示,利拉鲁肽及与其结构上紧密相关的代谢产物在乳汁中含量很低。大鼠哺乳期可见给药相关的新生鼠生长减慢。由于缺少相关经验,本品不得在哺乳期内使用。
在体外研究中已经证实,利拉鲁肽和其他活性物质之间发生与细胞色素 P450 和血浆蛋白结合有关的药代动力学相互作用的可能性极低。
利拉鲁肽对胃排空的轻度延迟可能会影响同时使用的口服药物的吸收。相互作用研究并未显示药物的吸收出现了任何与临床相关的延迟,因此无需调整剂量。少数经本品治疗的患者至少报告了 1 次严重腹泻事件。腹泻可能会影响同时使用的口服药物的吸收。
华法林和其他香豆素衍生物
尚未进行任何药物相互作用研究。不能排除本品与溶解性较差或治疗指数较窄的活性成分(如华法林)发生具有临床意义的相互作用的可能性。接受华法林或其他香豆素衍生物治疗的患者开始接受本品治疗后,推荐进行更为频繁的 INR(国际标准化比值)监测。
扑热息痛
利拉鲁肽不会改变扑热息痛单次给药 1000 mg 之后的总体暴露。扑热息痛的峰浓度(C<sub>max</sub>)降低了 31%,而达峰时间(t<sub>max</sub>)中位数延迟了 15 分钟。与扑热息痛联用时不需要进行剂量调整。
阿托伐他汀
利拉鲁肽对阿托伐他汀单次给药 40 mg 之后的总体暴露没有产生具有临床意义的改变。因此,阿托伐他汀与本品联用时不需要进行剂量调整。在利拉鲁肽的作用下,阿托伐他汀的峰浓度(C<sub>max</sub>)降低了 38%,而中位达峰时间(t<sub>max</sub>)从 1 小时延长至 3 小时。
灰黄霉素
利拉鲁肽不会改变灰黄霉素单次给药 500 mg 之后的总体暴露。灰黄霉素的峰浓度(C<sub>max</sub>)增加了 37%,而达峰时间(t<sub>max</sub>)中位数未发生变化。灰黄霉素和其他低溶解度和高渗透性的药物与本品联用均不需要进行剂量调整。
地高辛
单次给予地高辛 1 mg 同时合用利拉鲁肽可使地高辛的 AUC 降低 16%,C<sub>max</sub>降低 31%。t<sub>max</sub>从 1 小时延长至 1.5 小时。基于上述结果,无需调整地高辛给药剂量。
赖诺普利
单次给予赖诺普利 20 mg 同时合用利拉鲁肽可使赖诺普利的 AUC 降低 15%,C<sub>max</sub>降低 27%。达到赖诺普利峰浓度的中位 t<sub>max</sub>从 6 小时延长至 8 小时。基于上述结果,无需调整赖诺普利给药剂量。
口服避孕药
单次给予一种口服避孕药之后,利拉鲁肽分别使乙炔雌二醇和左炔诺孕酮的峰浓度(C<sub>max</sub>)降低了 12% 和 13%。利拉鲁肽使两种成份的达峰时间(t<sub>max</sub>)皆延长了 1.5 小时。对乙炔雌二醇或左炔诺孕酮的总体暴露没有产生具有临床意义的影响。因此,联用利拉鲁肽预期不会影响口服避孕药的避孕效果。
胰岛素
尚未对本品与胰岛素联用进行评价。
配伍禁忌
添加至本品的物质可能会导致利拉鲁肽的降解。在未进行配伍禁忌研究的情况下,本品不得与其他药品混合。
丹麦诺和诺德公司 Novo Nordisk A/S

86978997002263;86907103000077;86978997002256
利拉鲁肽是一种酰化人胰高糖素样肽-1(GLP-1)受体激动剂,其 97% 的氨基酸序列与内源性人 GLP-1(7-37)同源。GLP-1(7-37)占血液中所有内源性 GLP-1 的 20% 以下。与 GLP-1(7-37)相似,利拉鲁肽可活化 GLP-1 受体,GLP-1 受体是一类膜结合细胞表面受体,在胰腺β细胞中通过刺激性 G 蛋白 Gs,与腺苷酸环化酶偶联。当葡萄糖浓度升高时,利拉鲁肽可以增加细胞内环磷腺苷(cAMP),从而导致胰岛素释放。当血糖浓度下降并趋于正常时,胰岛素分泌减少。利拉鲁肽还可以葡萄糖依赖性地减少胰高糖素分泌。血糖水平降低的机制还涉及胃排空延迟。
遗传毒性
利拉鲁肽 Ames 试验、人外周血淋巴细胞染色体畸变试验、大鼠微核试验结果均为阴性。
生殖毒性
雄性大鼠在交配前 4 周和交配期间,皮下注射利拉鲁肽 0.1、0.25 和 1.0 mg/kg/d,在 1.0 mg/kg/d 剂量下,雄性动物生育力未受到直接的不良影响,根据血浆 AUC 计算,该剂量产生的全身暴露约为最大推荐人用剂量(MRHD)下人暴露的 11 倍。
雌性大鼠交配前 2 周至妊娠第 17 天,皮下注射利拉鲁肽 0.1、0.25 和 1.0 mg/kg/d,根据血浆 AUC 计算,3 个剂量产生的全身暴露约分别为 MRHD 下人暴露的 0.8、3 和 11 倍。在 1 mg/kg/d 剂量组中,早期胚胎死亡数略有增加。所有剂量下均可见胎仔异常以及肾脏和血管变异、颅骨不规则骨化和骨化过度完全状态。在 1.0 mg/kg/d 剂量下可见花斑状肝脏和轻微的肋骨扭结。发生率超过同期和历史对照的胎仔畸形为:0.1 mg/kg/d 剂量下口咽部畸形和/或喉开口处狭窄,0.1 和 0.25 mg/kg/d 剂量下脐疝。
兔妊娠第 6 天至第 18 天,皮下注射利拉鲁肽 0.01、0.025 和 0.05 mg/kg/d,根据血浆 AUC 计算,妊娠兔的全身暴露小于 MRHD 时的人暴露。在所有剂量下,胎仔体重降低,严重胎仔异常的总发生率呈剂量依赖性增加。在 0.01 mg/kg/d(肾脏、肩胛骨)、 ≥ 0.01 mg/kg/d(眼、前肢)、0.025 mg/kg/d(脑、尾和骶椎、大血管和心脏、脐)、 ≥ 0.025 mg/kg/d(胸骨)和 0.05 mg/kg/d(顶骨、大血管)剂量下,畸形的发生率超过同期和历史对照。不规则骨化和/或骨骼异常见于颅骨和颌骨、椎骨和肋骨、胸骨、骨盆、尾骨和肩胛骨;还可见剂量依赖性轻微骨骼变异。内脏异常见于血管、肺、肝脏和食管。所有给药组可见胆囊双叶或分叉,但对照组未见类似情况。
雌性大鼠在妊娠第 6 天至断乳或第 24 天终止哺乳期间,皮下注射利拉鲁肽 0.1、0.25 和 1.0 mg/kg/d,根据血浆 AUC 计算,全身暴露约分别为 MRHD 时人暴露的 0.8、3 和 11 倍。大多数给药组动物分娩期轻微延迟。给药组新生幼仔的组平均体重小于对照组。1 mg/kg/d 剂量组分娩的雌性大鼠出现血痂和激动行为。给药组 F2 子代大鼠出生至出生后第 14 天的平均体重低于对照组,但各组差异均未达到统计学意义。
致癌性
CD-1 小鼠 104 周致癌性试验中,皮下注射给予利拉鲁肽 0.03、0.2、1.0 和 3.0 mg/kg/d,根据血浆 AUC 计算,全身暴露量分别为 MRHD 时人暴露量的 0.2、2、10 和 45 倍。1.0 和 3.0 mg/kg/d 剂量组可见良性甲状腺 C 细胞腺瘤,发生率呈剂量依赖性增加,雄性小鼠发生率分别为 13% 和 19%,雌性小鼠分别为 6% 和 20%。对照组或 0.03 和 0.2 mg/kg/d 剂量组未见上述 C 细胞腺瘤。3.0 mg/kg/d 剂量组 3% 的雌性动物出现给药相关的恶性 C 细胞癌。在小鼠致癌性试验中甲状腺 C 细胞肿瘤为罕见。3 mg/kg/d 剂量组雄性动物给药部位皮肤和皮下组织出现给药相关的纤维肉瘤发生率增加。这些纤维肉瘤为注射部位药物局部浓度过高所致;临床使用的制剂中利拉鲁肽浓度(6 mg/mL)为上述 3 mg/kg/d 剂量组给药所用制剂浓度(0.6 mg/mL)的 10 倍。
SD 大鼠 104 周致癌性试验中,皮下注射给药利拉鲁肽 0.075、0.25 和 0.75 mg/kg/d,根据血浆 AUC 计算,全身暴露量分别为 MRHD 时人暴露量的 0.5、2 和 8 倍。雄性大鼠给予利拉鲁肽 0.25 和 0.75 mg/kg/d,良性甲状腺 C 细胞腺瘤发生率增加,与给药相关;各组雄性动物发生率分别为 12%、16%、42% 和 46%。雌性动物发生率分别为 10%、27%、33% 和 56%。利拉鲁肽各给药组雄性动物中,可见恶性甲状腺 C 细胞癌发生率增加,各组发生率分别为 2%、8%、6% 和 14%;0.25 和 0.75 mg/kg/d 剂量组雌性动物也可见发生率增加,分别为 4% 和 6%。在大鼠致癌性试验中甲状腺 C 细胞癌为罕见。
小鼠试验结果显示,利拉鲁肽引起的 C 细胞增生依赖于 GLP-1 受体,未激活甲状腺 C 细胞的原癌基因转染重排(RET)。
小鼠和大鼠中甲状腺 C 细胞肿瘤与人类的相关性尚未明确,尚无法通过临床研究或非临床研究确定。
药效学效应
利拉鲁肽的作用持续时间为 24 小时,能够通过降低 2 型糖尿病患者的空腹及餐后血糖而改善血糖控制。
在 2 型糖尿病患者中,单次给予利拉鲁肽可以观察到胰岛素分泌率以葡萄糖浓度依赖的模式增加(图 1)。

临床疗效和安全性
改善血糖控制以及降低心血管疾病发病率和死亡率都是 2 型糖尿病治疗的重要部分。
5 项双盲、随机、对照 3a 期临床试验对本品控制血糖的效果进行了评价。与安慰剂相比,本品治疗可使糖化血红蛋白 A1c(HbA1c)、空腹血糖和餐后血糖产生具有临床和统计学意义的改善。
这些研究共入选了 3978 例接受治疗的 2 型糖尿病患者(2501 例患者接受了本品治疗),其中 53.7% 为男性,46.3% 为女性,797 例患者(508 例接受本品治疗)的年龄 ≥ 65 岁,而 113 例患者(66 例接受本品治疗)的年龄 ≥ 75 岁。
此外,在 1901 例患者中对本品进行了另外 5 项临床试验,包括 4 项非盲、随机、对照临床试验(分别包括 464、658、323 和 177 例受试者)以及一项在 2 型糖尿病合并中度肾功能受损的受试者中进行的双盲、随机、对照临床试验(279 例患者)。
在 9340 例具有心血管高危风险的 2 型糖尿病患者中,进行了一项大型心血管结局试验(LEADER 试验)。 血糖控制
表 2 是一项为期 52 周关于本品单药治疗经过饮食和运动或一种口服抗糖尿病药物(OAD)血糖控制不佳受试者的临床试验结果。

在持续 26 周的试验中,与安慰剂相比,本品与二甲双胍、格列美脲或二甲双胍加罗格列酮联用可使患者的 HbA1c出现持续的降低,并且具有统计学意义(p<0.0001)(见表 3 和表 4)。




与格列美脲相比,本品单药治疗 52 周可使 HbA1c显著降低(p<0.0014)并且作用持久。

基线 HbA1c超过 9.5% 的患者接受本品单药治疗后 HbA1c平均降低了 2.1%,而在联合用药研究中接受本品的患者 HbA1c 的平均降低程度为 1.1-2.5%。
在肾功能受损患者中的使用
一项在国外 2 型糖尿病合并中度肾功能受损患者中开展的双盲试验,评估了在胰岛素和/或口服降糖药物治疗基础上,加用 1.8 mg 利拉鲁肽相较于安慰剂的有效性和安全性。结果显示,26 周治疗后,利拉鲁肽在降低 HbA1c 方面的效果优于安慰剂(-1.05% 相较-0.38%)。
利拉鲁肽治疗组与安慰剂组相比,达到 HbA1c<7% 的患者比例明显更高(52.8% 相较 19.5%)。两组体重均出现下降:利拉鲁肽组-2.4 kg,安慰剂组-1.09 kg。两组发生低血糖的风险相当。利拉鲁肽的安全性特征与其他利拉鲁肽研究中观察到的安全性特征相似。
HbA1c 达标的患者比例
经过 52 周的治疗后,本品单药治疗达到 HbA1c<7% 的患者比例显著高于接受格列美脲治疗的患者,且差异具有统计学意义(p ≤ 0.0007)。经过 26 周的治疗后,接受本品与二甲双胍、格列美脲或二甲双胍加罗格列酮联合治疗的患者中达到 HbA1c ≤ 6.5% 的比例要显著高于接受上述药物单药治疗的患者,且差异具有统计学意义(p ≤ 0.0001)。
空腹血糖
本品单用或联合一种或两种口服抗糖尿病药物治疗后可使空腹血糖降低 13-43.5 mg/dL(0.72-2.42 mmol/L)。这种降低在治疗的前两周内即可观察到。
餐后血糖
本品可使三餐后血糖均降低,降幅为 31-49 mg/dL(1.68-2.71 mmol/L)。
β细胞功能
根据稳态模型β细胞功能指数(HOMA-B)和胰岛素原/胰岛素的比值等检测方法,有关临床试验结果提示,本品可以改善β细胞功能。在使用本品治疗 52 周后,在 2 型糖尿病患者亚组中(N = 29)证实其可以改善第 1 和第 2 时相的胰岛素分泌。
体重
本品单药治疗 52 周后可以观察到持续的体重减轻。
本品与二甲双胍、二甲双胍 + 格列美脲或二甲双胍 + 罗格列酮联用时在试验期间观察到 1.0 kg 至 2.8 kg 的体重减轻。
基线时体重指数(BMI)越大的患者,体重减轻的程度越大。
心血管评估
对纳入了 5607 例患者(3651 例暴露于利拉鲁肽)的所有 II 期和 III 期中长期试验(持续 26 至 100 周)中的严重主要心血管不良事件(心血管死亡、心肌梗死、卒中)进行了事后分析,结果显示,与对照药(二甲双胍、格列美脲、罗格列酮、甘精胰岛素、安慰剂)相比,利拉鲁肽组的心血管风险复合终点未见增高(发生率为 0.75,95%Cl:0.35 至 1.63)。
2型糖尿病伴动脉粥样硬化性心血管疾病患者的心血管结局试验
LEADER 试验(NCT01179048)是一项多国家、多中心、安慰剂对照、双盲试验。在这项研究中,9340 例血糖控制不佳的 2 型糖尿病伴动脉粥样硬化性心血管疾病患者被随机分配接受诺和力 18 mg 或安慰剂治疗,中位持续时间为 3.5 年。这项研究比较了诺和力和安慰剂分别联合 2 型糖尿病标准治疗时的主要不良心血管事件风险。主要终点为从随机化至首次发生任何主要心血管不良事件(MACE)的时间,包括心血管死亡、非致死性心肌梗死或非致死性卒中。
入组人群为:年龄 ≥ 50 岁且确诊有稳定的心血管、脑血管、外周动脉疾病、慢性肾病或 NYHA 心功能分级为 II 级和 III 级的心力衰竭(占入组人群的 80%)或年龄 ≥ 60 岁且伴有其他特定的心血管疾病风险因素(占入组人群的 20%)的患者。
基线时,组间人口统计学和疾病特征分布均衡。平均年龄为 64 岁,人群中 64.3% 为男性,77.5% 为高加索人,10.0% 为亚洲人,8.3% 为黑人。在这项研究中,12.1% 的人群为西班牙裔或拉丁裔。2 型糖尿病的平均病程为 12.8 年,平均 HbA<sub>1c</sub>为 8.7%,平均 BMI 为 32.5 kg/m<sup>2</sup>。31% 的随机受试者有心肌梗死病史,39% 有血流重建术既往史,11% 有缺血性卒中病史,9% 有症状性冠状动脉疾病史,26% 有无症状心肌缺血,14% 为纽约心脏病学会(NYHA)心功能分级 I 级至 III 级的心力衰竭。基线时的平均 eGFR 为 79 mL/min/1.73m<sup>2</sup>;41.8% 的患者伴轻度肾功能损害(eGFR60-90 mL/min/1.73m<sup>2</sup>),20.7% 的患者伴中度肾功能损害(eGFR30-60 mL/min/1.73m<sup>2</sup>),2.4% 的患者伴重度肾功能损害(eGFR<30 mL/min/1.73m2)。
基线时,患者糖尿病的治疗情况包括:仅通过饮食和运动控制(3.9%)、仅使用口服降糖药物(51.5%)、使用口服降糖药物和胰岛素(36.7%)或仅使用胰岛素(7.9%)。基线和试验中使用的最常见的背景降糖药物为二甲双胍、磺脲类药物和胰岛素。方案排除了 DPP4-抑制剂和其他 GLP-1 受体激动剂的使用,SGLT-2 抑制剂或是未获批或是未广泛使用。在基线时,对于心血管疾病和风险因素的治疗用药包括非利尿性抗高血压药(92.4%)、利尿剂(41.8%)、他汀类药物(72.1%)和血小板聚集抑制剂(66.8%)。试验期间,研究者可以调整抗糖尿病药物和心血管药物,以达到当地血糖、血脂和血压的标准治疗目标,并根据当地治疗指南管理正在从急性冠脉综合征或卒中事件中恢复的患者。
对于主要分析,使用 Cox 比例风险模型对 MACE 风险比预先规定的风险界值 1.3 进行非劣效性检验,并在证明非劣效性的情况下对 MACE 的优效性进行检验。在多项检验中控制 1 类错误。
利拉鲁肽显著降低了 MACE 的发生风险。至首次发生 MACE 的时间的估计风险比(95%Cl)为 0.87(0.78,0.97)(图 3 及表 6)。


试验中 99.7% 的受试者可获得生命状态信息。LEADER 试验期间共记录了 828 例死亡。大多数死亡归类为心血管死亡,治疗组之间的非心血管死亡率均衡(接受诺和力治疗的患者为 3.5%,接受安慰剂治疗的患者为 3.6%)。与安慰剂组相比,诺和力组至全因死亡时间的估计风险比为 0.85(0.74,0.97)。
利拉鲁肽同时显著降低了扩展的 MACE(MACE、导致住院的不稳定型心绞痛、冠状动脉血流重建或因心力衰竭住院)和其他次要终点的风险(图 4)。

在标准治疗的基础上,与安慰剂相比,观察到利拉鲁肽组 HbA1c 从基线至第 36 个月显著且持续降低(-1.16% 与-0.77%;估计的治疗差异[ETD]-0.40%[-0.45;-0.34])。在基线时未接受胰岛素治疗的患者中,与安慰剂相比,利拉鲁肽组对胰岛素强化治疗的需求降低了 48%(HR 0.52[0.48;0.57])。
血压和心率
在 3a 期试验期间,利拉鲁肽组收缩压相对基线平均降低 2.3-6.7 mmHg,活性对照药组降低 1.9-4.5 mmHg。在长期临床试验(包括 LEADER)中,已经观察到利拉鲁肽组心率相对基线平均增加 2-3 次/分钟。在 LEADER 试验中,未观察到心率增加对心血管事件风险的长期临床影响。
微血管评估
在 LEADER 试验中,微血管事件由肾病和视网膜病变结局组成。利拉鲁肽与安慰剂至首次微血管事件发生时间的分析结果为 HR 0.84[0.73,0.97]。利拉鲁肽与安慰剂至首次肾病事件发生时间的 HR 为 0.78[0.67,0.92],至首次视网膜病变事件发生时间的 HR 为 1.15[0.87,1.52]。
在 LEADER 试验中,共纳入中国受试者 92 例(利拉鲁肽组 48 例,安慰剂组 44 例),平均年龄为 63.5 岁,平均 BMI 为 26.9 kg/m<sup>2</sup>,糖尿病的平均病程为 14.8 年。包括轻度(n = 25)、中度(n = 5)肾功能受损的患者。
主要研究终点为至首起 MACE 事件的时间,在接受利拉鲁肽与安慰剂治疗的中国受试者中,共报告了 9 例 MACE 事件,其中利拉鲁肽组有 4 例患者(8.33%),安慰剂组有 5 例患者(11.36%)。利拉鲁肽组与安慰剂组至首次 MACE 事件发生时间的分析结果为 HR 0.708[0.190;2.637]95%Cl,这与全部受试者的结果一致。中国受试者中观察到的心血管安全性的结果总体上也与全部受试者中的结果一致。具体临床试验结果详见表 7。

吸收
利拉鲁肽经皮下注射后的吸收比较缓慢,在给药后 8-12 小时达到最大浓度。单次皮下注射利拉鲁肽 0.6 mg 之后,利拉鲁肽的最大浓度估计值为 9.4 nmol/L。在 1.8 mg 的利拉鲁肽剂量水平下,利拉鲁肽的平均稳态浓度(AUC<sub>T/24</sub>)达到约 34 nmol/L。利拉鲁肽的暴露程度随剂量成比例增加。单次给予利拉鲁肽,药时曲线下面积(AUC)的个体内变异系数为 11%。
利拉鲁肽皮下注射后的绝对生物利用度约为 55%。
分布
皮下注射后的表观分布容积为 11-17L。
利拉鲁肽静脉注射后的平均分布容积为 0.07L/kg。利拉鲁肽可与血浆蛋白广泛结合(>98%)。
代谢
单次给予健康受试者放射标记的[3H]-利拉鲁肽的 24 小时内,血浆中的主要成分为利拉鲁肽原型药物。检测到两种少量血浆代谢产物(分别为总血浆放射性暴露的 ≤ 9% 和 ≤ 5%)。利拉鲁肽以一种与大分子蛋白类似的方式进行代谢,尚无特定器官被确定为主要的消除途径。
消除
[3H]-利拉鲁肽给药后,在尿液和粪便中没有检测到完整的利拉鲁肽。所给予的放射性中仅有少部分作为利拉鲁肽相关的代谢产物经尿液或粪便排泄(分别是 6% 和 5%)。
尿液和粪便中的放射性主要在前 6-8 天内排泄,分别对应于三种少量的代谢产物。
利拉鲁肽单次皮下注射后的平均清除率约为 1.2L/小时,消除半衰期约为 13 小时。
特殊人群
性别:对男性及女性患者的群体药代动力学数据分析结果以及一项在健康受试者中进行的药代动力学研究结果显示,性别不会对利拉鲁肽的药代动力学产生具有临床意义的影响。
种族来源:一项包含白人、黑人、亚洲人和西班牙人受试者的群体药代动力学分析的结果显示,种族差异不会对利拉鲁肽的药代动力学产生与临床相关的影响。
肥胖:群体药代动力学分析提示,体重指数(BMI)不会对利拉鲁肽的药代动力学产生显著影响。
肝功能受损:一项单次给药临床试验评价了利拉鲁肽在不同程度肝功能受损受试者中的药代动力学。与健康受试者相比,轻至中度肝功能受损受试者的利拉鲁肽暴露降低了 13-23%。重度肝功能受损(Child Pugh 评分 > 9)受试者的利拉鲁肽暴露显著降低(44%)。
肾功能受损:与肾功能正常的受试者相比,肾功能受损受试者的利拉鲁肽暴露降低。轻度(肌酐清除率,CrCl 50-80 mL/min)、中度(CrCl 30-50 mL/min)以及重度(CrCl<30 mL/min)肾功能受损和需要透析的终末期肾病受试者的利拉鲁肽暴露分别降低了 33%、14%、27% 和 28%。与此类似,在一项为期 26 周的临床试验中,2 型糖尿病合并中度肾功能受损(肌酐清除率 30-59 mL/min,患者的利拉鲁肽暴露量与一项入选肾功能正常或轻度肾功能受损的 2 型糖尿病患者的试验相比降低了 26%。
注射剂
3ml:18mg(预填充注射笔)
本品为可调节剂量、一次性预填充注射笔,由笔型注射器和装有 3 mL 液体的笔芯组成。笔芯由 1 型玻璃制成,内有一个活塞(溴丁基橡胶),并由一个橡胶塞(溴丁基橡胶聚异戊二烯橡胶)密封。 笔型注射器由聚烯烃和聚缩醛制成。 每支笔含有 3 mL 溶液,可以进行 30 次 0.6 mg、15 次 1.2 mg 或 10 次 1.8 mg 注射。 包装规格:每盒 1 支。每盒 2 支。
本品应冷藏于 2 ℃ -8 ℃ 冰箱中(勿接近冰箱的冷冻室)。不可冷冻。 首次使用后,应在 30 ℃ 以下贮藏或冷藏在 2 ℃ -8 ℃ 冰箱中,不可冷冻;首次使用后的有效期为 1 个月。 盖上笔帽避光保存。 应当告知患者在每次注射后按照当地的要求丢弃注射针头,同时,贮藏本注射笔时切勿带有针头。这可以避免污染、感染和渗漏,同时能确保给药准确。
278
30 个月。
S20160005;国药准字J20160037;S20160004

微信扫一扫,关注医库最新动态