百悦泽 Brukinsa说明书
Zanubrutinib Capsules
泽布替尼
本品活性成份为泽布替尼。
化学名称:(S)-7-[4-(1- 丙烯酰基哌啶基 )]-2-(4- 苯氧基苯基 )-4,5,6,7- 四氢吡唑并 [1,5-a] 嘧啶 -3- 甲酰胺
化学结构式:

分子式:C27H29N5O3
分子量:471.55
泽布替尼胶囊为白色至类白色硬胶囊,内容物为白色至类白色粉末。
本品适用于:1) 既往至少接受过一种治疗的成人套细胞淋巴瘤(MCL)患者。2) 既往至少接受过一种治疗的成人慢性淋巴细胞白血病(CLL)/ 小淋巴细胞淋巴瘤(SLL)患者。分别基于一项单臂临床试验的客观缓解率结果附条件批准上述适应症,完全批准将取决于正在开展中的确证性随机对照临床试验结果。3)既往至少接受过一种治疗的成人华氏巨球蛋白血症(WM)患者。基于一项单臂临床试验的主要缓解率结果附条件批准上述适应症,完全批准将取决于正在开展中的确证性随机对照临床试验结果。
本品须在有血液系统肿瘤治疗经验医生的指导下用药。应口服给药,每天的用药时间大致固定。应用水送服整粒胶囊,可在饭前或饭后服用。请勿打开、弄破或咀嚼胶囊。如果未在计划时间服用本品,患者应在相邻服药间隔至少 8 小时基础上尽快服用,并在后续恢复正常用药计划。请勿额外服用本品以弥补漏服剂量。推荐剂量为每次160mg(2粒80mg胶囊),口服,每日两次,直到发生疾病进展或出现不可耐受的毒性。与CYP3A抑制剂或诱导剂联合给药时的剂量调整:与CYP3A抑制剂或诱导剂联合给药时的剂量调整见表 1。表 1:与 CYP3A 抑制剂或诱导剂联合给药时的剂量调整
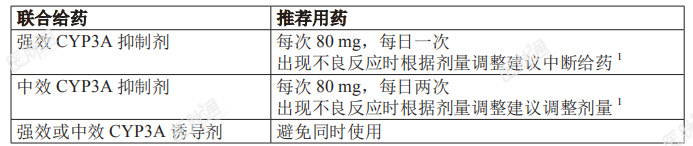
参见出现不良反应时的剂量调整部分:停止使用 CYP3A 抑制剂后,恢复本品剂量调整前用量推荐剂量及肝功能损伤患者剂量调整部分。出现不良反应时的剂量调整:剂量调整建议见表 2。表 2:建议按如下所述进行剂量调整
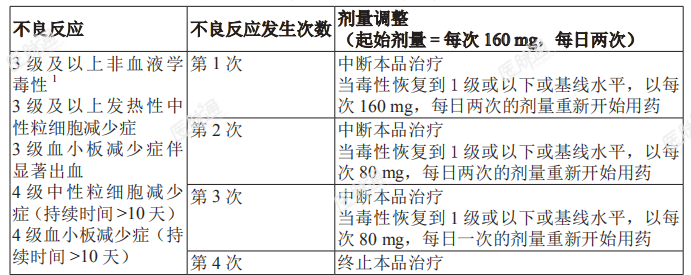
通过口服药物得到充分控制的高血压或无症状的实验室检查异常除外;提示存在肝或肾功能障碍的实验室检查异常不视为无症状的实验室检查异常。无症状的淋巴细胞增多不应视为不良反应,出现此事件的患者应继续服用本品。特殊人群用药:肝功能损伤:轻度至中度肝损伤患者不建议进行剂量调整。重度肝损伤患者推荐剂量是每次80mg(1粒80mg胶囊),口服,每日两次。肾功能损伤:肾功能损伤患者不建议进行剂量调整。重度肾功能损伤(肌酐清除率 < 30mL/min)或透析患者使用本品需监测相关不良反应。老年用药:
老年患者无需进行剂量调整。儿童用药:本品在儿童患者中的安全性和有效性尚未确立。
以下不良反应的详细内容请参见说明书。
.出血
.血细胞减少症
.感染
.乙肝病毒再激活
.第二原发恶性肿瘤
.心律失常
.肿瘤溶解综合征
临床试验经验:由于临床试验是在各种不同条件下进行的,在一项药物的临床试验中观察到的不良反应发生率不能直接与另一项药物的临床试验中的发生率进行比较,并且可能并不反映实践中观察到的发生率。安全性特性总结:安全性特性基于六项单药临床试验中779例接受泽布替尼治疗的B细胞恶性肿瘤患者的汇总数据,这些临床试验包括一项1期临床研究(BGB-3111-1002)、一项1/2期临床研究(BGB-3111-AU-003)、三项2期研究(BGB-3111-205、BGB-3111-206、BGB-3111-210)以及一项3期临床研究(BGB-3111-302)。根据六项研究的汇总数据,十分常见的不良反应(≥20%)为中性粒细胞减少症、血小板减少症、上呼吸道感染、贫血、皮疹、骨骼肌肉疼痛以及腹泻。常见的3级或以上不良反应(≥5%)为中性粒细胞减少症、血小板减少症、感染性肺炎以及贫血。严重不良反应的发生率为18.0%,常见为感染性肺(10.0%)。779例接受泽布替尼治疗的患者中有47例(6.0%)患者因不良反应终止治疗,导致治疗终止的常见不良反应为感染性肺炎(1.3%)。236例(30.3%)的患者因不良反应中断给药,导致中断给药的常见不良反应为感染性肺炎(5.9%)和中性粒细胞减少症(5.4%)。41例(5.3%)的患者出现导致剂量降低的不良反应,导致剂量降低的常见不良反应为腹泻(1.0%)。下表3所示为六项临床研究中报告的泽布替尼单药治疗过程中出现的不良反应。表3:接受泽布替尼治疗患者出现的不良反应&
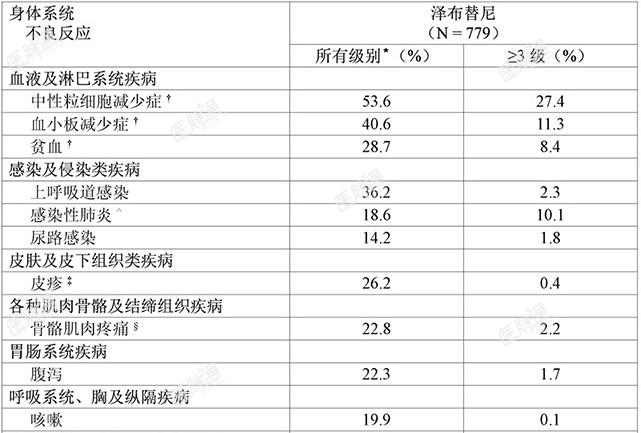
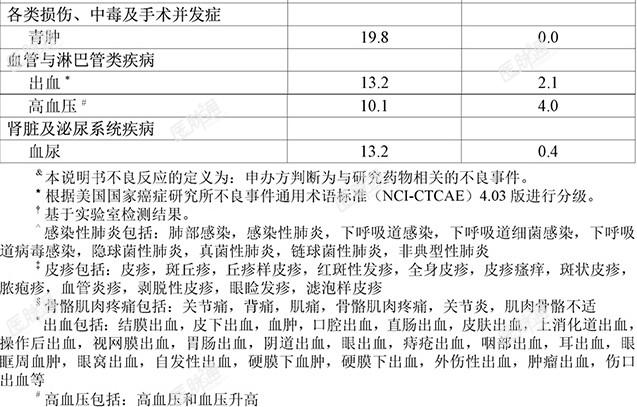
本品禁用于已经对泽布替尼或辅料超敏(如速发过敏和类速发过敏反应)的患者。
出血:在接受本品单药治疗的血液系统恶性肿瘤患者中,曾发生严重出血和致死性出血事件。3.1%的患者发生严重出血事件,其中包括血尿和上消化道出血(各0.4%)。3.5%的患者发生3级或更高级别的出血事件,其中包括血尿(0.4%)、硬膜下出血(0.3%)、胃肠出血(0.3%)和胸腔积血(0.3%)。52.8%的患者发生任意级别的出血事件,以紫癜/瘀点/青肿和血尿常见(>10%)。分别有0.6%、4.6%和1.3%的患者因出血事件降低本品剂量、暂停给药和终止治疗。出血事件的发生机制尚不清楚。本品可能会增加接受抗血小板或抗凝治疗患者的出血风险,应监测患者的出|亚血迹象。需接受手术的患者,应根据手术类型和出血的风险,进行风险获益评估,考虑在术前和术后暂停本品3-7天。如发生与治疗有关的3级或3级以上的出血或任何级别的颅内出血时,应永久终止本品治疗。感染:在接受本品单药治疗的血液系统恶性肿瘤患者中有致死性严重感染(包括细菌、病毒或真菌)和机会性感染的报告。有26.3%患者发生3级或以上级别的感|职染事件,其中最常见的是感染性肺炎(10.1%)。分别有1.5%、16.6%和2.7%的患者发生导致降低本品剂量、暂停给药和治疗终止的感染事件。对感染高危患者,应考虑对单纯疱疹病毒、耶氏肺孢子虫肺炎和其他感染进行预防治疗。监测和评估患者是否出现发热或其他感染的症状和体征,并给予相应治疗。乙肝病毒(HBV)再激活:在本品单药的临床试验中有乙型肝炎病毒再激活(1.2%)报告。在这些临床试验中,活动性乙型肝炎患者已除外。本品对于乙型肝炎病毒再激活的影响尚不清楚。应在使用本品前明确乙型肝炎病毒状态。若患者目前或既往有乙型肝炎病毒感染,建议在开始本品治疗前咨询肝炎专科医师,并依据当地诊疗常规监测管理,以防止乙型肝炎复发。在临床试验中乙型肝炎核心抗体阳性患者须接受预防性抗乙型肝炎病毒治疗。血细胞减少症:基于实验室检测结果,在接受本品单药治疗的血液系统恶性肿瘤患者中,血细胞减少十分常见,表现为中性粒细胞减少、血小板减少和贫血,并常见有3级或4级血细胞减少症的报告。常见(≥1%)患者因血细胞减少症,包括中性粒细胞减少症(8.2%)、血小板减少症(2.3%)和贫血(1.2%)中断治疗;分别有1.3%、0.3%和0.1%患者因中性粒细胞减少症、血小板减少症和贫血降低本品剂量;偶见(<1%)患者因血细胞减少终止治疗。在治疗期间建议密切监测全血细胞计数,如发生血细胞减少,应根据临床需求给予对症治疗;必要时暂停用药,待相关血液学不良反应缓解至用药条件后再恢复用药。第二原发恶性肿瘤:同类产品有发生第二原发恶性肿瘤的报道。在接受本品单药治疗的血液系统恶性肿瘤患者中,有12.1%发生第二原发恶性肿瘤,最常见的是皮肤癌(8.1%)(其中包括基底细胞癌[4.5%]和皮肤鳞状细胞癌[2.8%])。建议患者做好防晒措施。心律失常:同类产品的临床试验和上市后观察中均报道过房颤、房扑及室性心动过速。在接受本品单药治疗的血液系统恶性肿瘤患者中,有2.2%的患者发生房颤或房扑事件,其中0.6%的患者为3级或以上级别事件;0.6%的患者发生室性期外收缩或室性心律失常,其中0.1%的患者为3级或以上级别事件。存在心脏风险因素、患有高血压和急性感染患者的风险可能会增加。在接受本品治疗期间,应定期监测患者是否发生心律失常,对出现心律不齐症状(如心悸、头晕、昏厥、胸部不适或新发呼吸困难)的患者进行临床评价,根|亚据指征要求患者接受心电图(ECG)检查。出现心律失常时应及时调整治疗。肿瘤溶解综合征:使用本品单药治疗时,尤其是在接受治疗的慢性淋巴细胞白血病(CLL)患者中,已有肿瘤溶解综合征个案报告。治疗前应评估风险(如高肿瘤负荷)并采取适当的预防措施。密切监测患者并予以适当的治疗。高血压:在接受本品单药治疗的血液系统恶性肿瘤患者中,有10.1%的患者发生高血压事件,其中4.0%为3级或者以上级别。无患者因高血压导致降低本品剂量或终止治疗。仅有1例(0.1%)患者因高血压暂停给药。应定期监测接受本品治疗患者的血压,并酌情使用新的降压药或调整原有降压药治疗。特殊人群:肝功能损伤:本品在重度肝损伤患者中的安全性尚未建立。重度肝损伤患者建议调整本品剂量。轻度至中度肝损伤患者不建议改变剂量。肝损伤患者接受本品治疗时应监测不良反应。肾功能损伤:本品经肾消除量极少。临床试验中入组了轻度和中度肾损伤患者,尚未在严重肾损伤患者或需要透析的肾损伤患者中评估本品的药代动力学特征。对于伴轻至中度肾功能损伤(肌酐清除率≥30mL/min,根据Cockcroft-Gault公式进行估算)的患者,不建议进行剂量调整。重度肾功能损伤(肌酐清除率<30mL/min)或透析患者使用本品需监测相关不良反应。育龄女性和男性:妊娠检测:在本品治疗开始前,需对有生育能力的女性进行妊娠状态检查。避孕:本品可能产生胚胎-胎儿毒性。建议有生育能力的女性在本品治疗期间及治疗结束后1周内采取高效避孕措施。如果在怀孕期间服用本品或服用本品期间怀孕,应明确告知患者本品可能对胎儿造成危害。
建议男性在本品治疗期间以及治疗结束后1周内采取高效避孕措施。对驾驶及操作机械能力的影响:尚未进行研究来评价本品对驾驶和操作机械能力的影响。其他:远离儿童放置。
在泽布替尼用药过量的处理方面尚无具体经验。本品无特定解毒剂。对于服用过量的患者,应进行密切监测并提供适当的支持性治疗。
本品 MCL(BGB-3111-206)和 CLL/SLL(BGB-3111-205)和WM(BGB-3111-210)关键临床试验中,≥ 65岁的患者分别占25.6%、34.1%和52.3%。对老年患者无需因为年龄因素进行剂量调整。
CYP3A 抑制剂对泽布替尼的作用

CYP3A 诱导剂对泽布替尼的作用

百济神州(苏州)生物科技有限公司

86982282000018
套细胞淋巴瘤(MCL):BGB-3111-206一项在中国开展的开放、多中心、单臂、2 期临床试验(BGB-3111-206)中,对泽布替尼治疗既往至少接受过一种治疗的 MCL 患者的安全性和有效性进行了评价。共纳入 86 例复发 / 难治性 MCL 中国患者,中位年龄为 61 岁(范围:34-75 岁),77.9%为男性。自确定诊断后的平均时间为36月,接受既往治疗的中位线数为2线(范围:1-4 线)。大多数患者疾病分期为 III/IV 期(16.3% 为 III 期,74.4% 为 IV 期);83.7% 的患者具有中 / 高风险 MIPI-b 评分,52.3% 的患者为难治性疾病,45.3% 的患者伴有骨髓受累,70.9% 的患者有结外病灶。入组患者接受本品每次 160 mg,每日两次口服。中位随访时间为 18.4 月(范围:0.3-23.5 月)。由独立审查委员会(IRC)基于 PET-CT 根据修订的恶性淋巴瘤缓解标准评估的疗效结果参见表 4。表 4:复发 / 难治性 MCL 患者疗效结果(试验 BGB-3111-206)
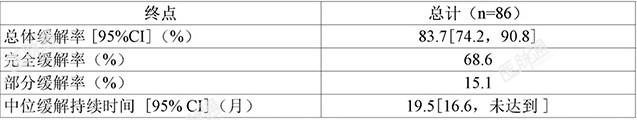
基于上述Ⅱ期单臂临床试验的结果附条件批准本适应症。该适应症的完全批准将取决于正在进行的确证性试验证实本品的临床获益。慢性淋巴细胞白血病 / 小淋巴细胞淋巴瘤(CLL/SLL)BGB-3111-205一项在中国开展的开放、多中心、单臂、2期临床试验(BGB-3111-205)中,对泽布替尼治疗既往至少接受过一种治疗的 CLL/SLL 患者的安全性和有效性进行了评价。共纳入91例确诊为复发 / 难治性 CLL/SLL 中国患者,90.1%为CLL患者,9.9% 为 SLL患者;中位年龄为61岁(范围:35-87 岁),57.1% 为男性;44.0%患者存在至少1个最长径≥ 5cm 的病灶,9.9% 患者存在长径≥ 10cm的病灶。22.0%的患者存在11q缺失,24.2%存在17p缺失或 TP53突变,56.0% 伴有未突变的IGHV。接受既往治疗的中位线数为1线(范围:1-9 线),79.1% 的患者末次治疗评估为难治。入组患者接受本品每次160mg,每日两次口服;中位随访时间15.1月(范围:0.8-21.2 月);由独立审查委员会(IRC)根据修订的 IWCLL 指导原则 (2008) 和恶性淋巴瘤缓解标准修订版对 CLL 患者和 SLL 患者进行肿瘤缓解评估的疗效结果见表 5。表 5:复发 / 难治性 CLL/SLL 患者疗效结果(试验 BGB-3111-205)
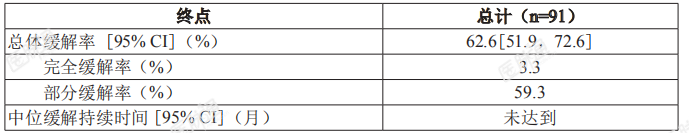
另有22.0%患者获得伴有淋巴细胞增多的部分缓解。基于上述Ⅱ期单臂临床试验的结果附条件批准本适应症。该适应症的完全批准将取决于正在进行的确证性试验证实本品的临床获益。华氏巨球蛋白血症(WM)
BGB-3111-210一项在中国开展的开放、多中心、单臂2期临床试验(BGB-3111-210)对泽布替尼治疗既往至少接受过一种治疗的WM患者的安全性和有效性进行了评价。共入组了44例中国患者且均至少接受过一次本品治疗。本研究患者的中位年龄为65岁(范围:41-83岁)。43.2%的总人群>65岁。27例患者(61.4%)为男性。自疾病进展至首次研究药物治疗的中位时间相对较短,为1.22年。根据WM国际预后评分系统,20例患者(45.5%)在研究入组时被评估为高危患者。患者有贫血倾向,中位血红蛋白水平为98g/L并且72.7%的患者在基线时患有髓外疾病。既往全身治疗中位方案数为2。70.5%的患者在既往最后一次全身治疗中未能实现轻微缓解或更好的缓解。入组患者接受本品每次160mg,每日两次口服。中位随访时间为14.9个月(范围:3.2-20.2个月)。由IRC根据修订的Owen2013评估的有效性结果见表6。
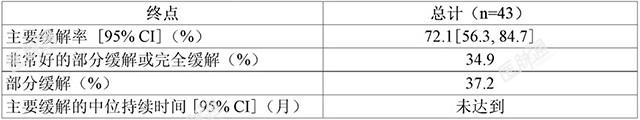
基于上述Ⅱ期单臂临床试验的结果附条件批准本适应症。该适应症的完全批准将取决于正在进行的确证性试验证实本品的临床获益。
胶囊剂
80mg
口服固体药用高密度聚乙烯瓶及高密度聚乙烯/聚丙烯儿童安全组合瓶盖系统。 60粒/瓶/盒;64粒/瓶/盒;120粒/瓶/盒。
密封,30℃以下保存。
2778.00
36个月。
国药准字H20200005

微信扫一扫,关注医库最新动态