罗圣全说明书
Entrectinib Capsules
恩曲替尼
本品活性成份为恩曲替尼。
化学名称:N-{5-[(3,5-二氟苯基)甲基]-1H-吲唑-3-基}-4-(4-甲基哌嗪-1-基)-2-[(四氢吡喃-4-基)氨基]苯甲酰胺
化学结构式:
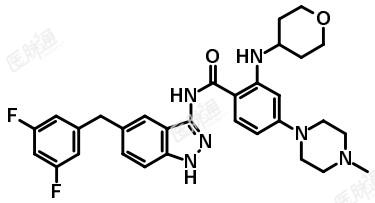
分子式:C31H34F2N6O2
分子量:560.64
辅料:无水乳糖,酒石酸,羟丙甲纤维素,交联聚维酮,微晶纤维素,胶态二氧化硅,硬脂酸镁。胶囊壳中含有羟丙甲纤维素,二氧化钛、氧化铁黄(100mg 规格)、日落黄(200mg 规格)。印墨中含有虫胶、丙二醇、浓氨溶液、靛蓝铝色锭。
本品为黄色或类黄色薄膜衣片,除去包衣后显淡黄色或黄色。
实体瘤:本品适用于符合下列条件的成人和12岁及以上儿童实体瘤患者,经充分验证的检测方法诊断为携带神经营养酪氨酸受体激酶(NTRK)融合基因且不包括已知获得性耐药突变,患有局部晚期、转移性疾病或手术切除可能导致严重并发症的患者,以及无满意替代治疗或既往治疗失败的患者。本适应症为基于替代终点获得附条件批准上市,暂未获得临床终点数据,有效性和安全性尚待上市后进一步确证。非小细胞肺癌(NSCLC):本品适用于ROS1阳性的局部晚期或转移性非小细胞肺癌(NSCLC)成人患者。
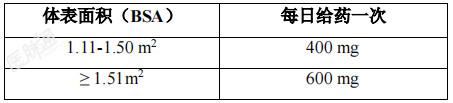
实体瘤:本品的治疗应由具有抗癌治疗经验的医生启动。在使用本品治疗之前,必须确定患者肿瘤样本中携带NTRK融合基因。应采用验证过的检测方法确定患者的NTRK融合基因状态。经医院或实验室的检测结果判断为携带NTRK融合基因的患者能接受本品治疗,并且应经罗氏公司指定的独立第三方进行一次审核,证实患者确具有NTRK融合基因可继续用药。非小细胞肺癌(NSCLC):需要采用经验证的检测方法,选择ROS1阳性局部晚期或转移性NSCLC患者。应在开始恩曲替尼治疗前确定ROS1阳性状态。推荐剂量:本品适用于口服给药。硬胶囊应整粒吞服,由于内容物较苦,因此不得打开或溶解后服药。本品可与或不与食物同服,但不应与葡萄柚或葡萄柚汁同服。成人:成人的推荐剂量为600mg,口服,每日一次。儿童患者:年满 12岁儿童患者的推荐剂量为300mg/m2,口服,每日一次(参见表 1)。表1: 儿童患者的推荐给药方案
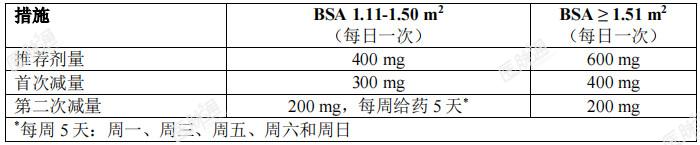
治疗持续时间:建议患者接受本品治疗直至疾病进展,或出现无法耐受的毒性。延误或漏服:如患者漏服,可补服,除非距离下一次服药不足12小时。如患者服用恩曲替尼后立即呕吐,患者可再次服药。剂量调整:管理不良事件时,可能需要暂时中断给药、降低剂量或停止恩曲替尼治疗,具体根据处方医师对患者安全或耐受性的评估而定。成人:根据耐受性,成人的恩曲替尼剂量可最多减量两次。表2提供了成人患者的通用剂量调整建议。如果患者不能耐受200mg每日一次的剂量,应永久停止恩曲替尼治疗。表2: 成人患者的减量方案


儿童患者:表3 提供了儿童患者的特殊减量建议。根据耐受性,年满12岁的儿童患者可最多减量两次。某些儿童患者需要采取间歇给药方案才能达到推荐的减量后的一周总剂量。如果患者不能耐受减量后的最低推荐剂量,应永久停止恩曲替尼治疗。表3: 儿童患者的剂量减量方案
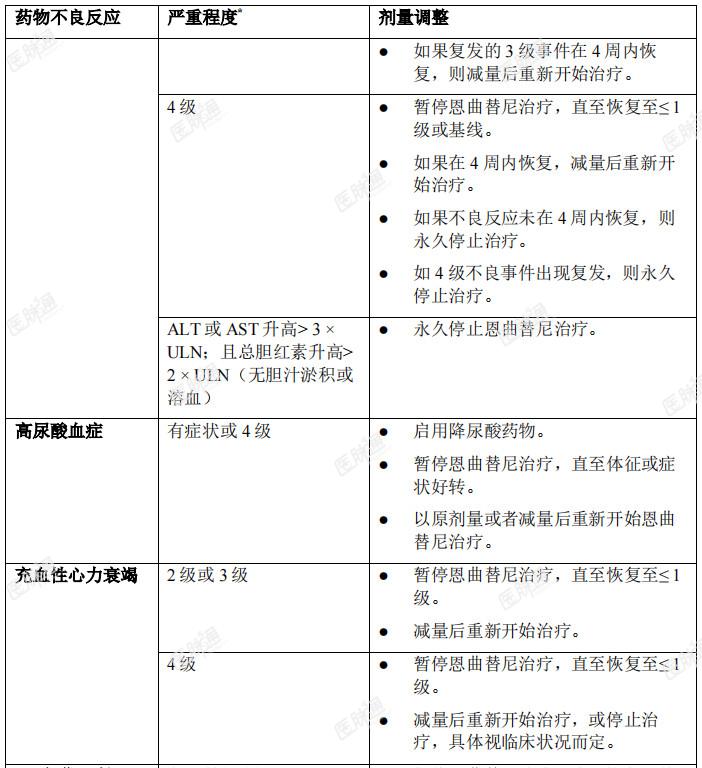
发生特定不良反应后的剂量调整:成人和儿童患者发生特定药物不良反应后的恩曲替尼剂量调整建议见表4。表4:成人和儿童患者发生特定药物不良反应后的推荐剂量调整方案
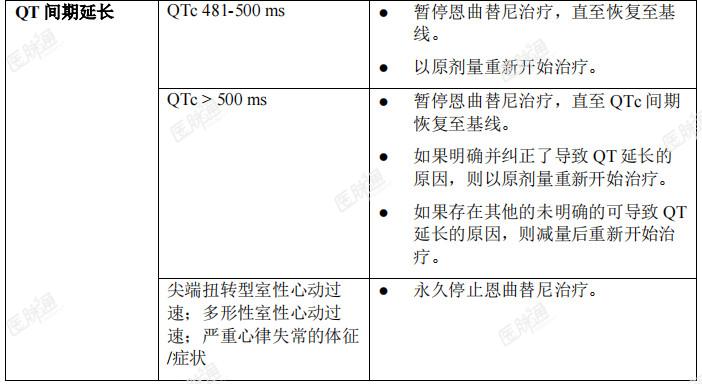

发生特殊药物相互作用后的剂量调整:CYP3A强效或中效抑制剂合并用药:成人:在成人患者中,应避免与强效或中效CYP3A抑制剂合并服用,或者合并服用时间应限制在14天以内。如果合并服用无法避免,则与强效CYP3A抑制剂合并服用时,应将恩曲替尼剂量降至100mg每日一次,与中效CYP3A抑制剂合并服用时,应将恩曲替尼剂量降至200mg每日一次。在停止合并服用的强效或中效CYP3A抑制剂后,可以恢复恩曲替尼至合并用药前的剂量。半衰期长的CYP3A4抑制剂可能需要洗脱期。儿童患者:在年满12岁的儿童患者中,应该避免合并服用强效或中效CYP3A抑制剂。CYP3A诱导剂合并用药:在成人和儿童患者中,应避免合并服用CYP3A诱导剂。特殊人群剂量说明:儿童:尚未确立恩曲替尼在12岁以下儿童中的安全性和有效性。老年人:年龄≥ 65岁的患者无需调整恩曲替尼剂量。肾功能不全:轻度或中度肾功能不全患者无需调整剂量。尚未在重度肾功能不全患者中研究恩曲替尼的安全性与有效性。然而,由于恩曲替尼的肾脏消除率可忽略不计,重度肾功能不全患者无需调整剂量。肝功能不全:对于轻度肝功能不全患者,不建议进行剂量调整。尚未在中重度肝功能不全患者中研究恩曲替尼的安全性与有效性。
安全性特征总结:最常见的不良反应(≥20%)为疲乏、便秘、味觉障碍、水肿、头晕、腹泻、恶心、感觉迟钝、呼吸困难、贫血、体重增加、血肌酐升高、疼痛、认知障碍、呕吐、咳嗽和发热。最常见的严重不良反应(≥2%)为肺部感染(5.2%)、呼吸困难(4.6%)、认知障碍(3.8%)、胸腔积液(3.0%)和骨折(2.4%)。4.6%的患者因不良反应永久停止治疗。不良反应汇总表:表5和表6总结了在3项成人临床试验(ALKA、STARTRK-1、STARTRK-2)和1项儿童临床试验(STARTRK-NG)中接受恩曲替尼治疗的成人和儿童患者发生的药物不良反应(ADR)。中位暴露时间为5.5个月。药物不良反应按MedDRA系统器官分类列示。采用的发生频率分类如下:
十分常见(≥1/10)、常见(≥1/100至<1/10)、少见(≥1/1,000至<1/100)、罕见(≥1/10,000至<1/1000)、十分罕见(<1/10,000)。在每个系统器官分类中,不良反应按发生率降序排列。表5:在临床试验中接受恩曲替尼治疗的成年和儿童患者发生的药物不良反应(N=504)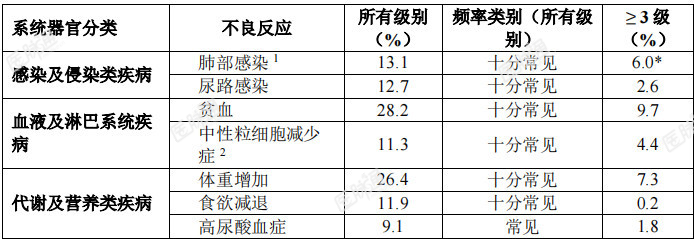
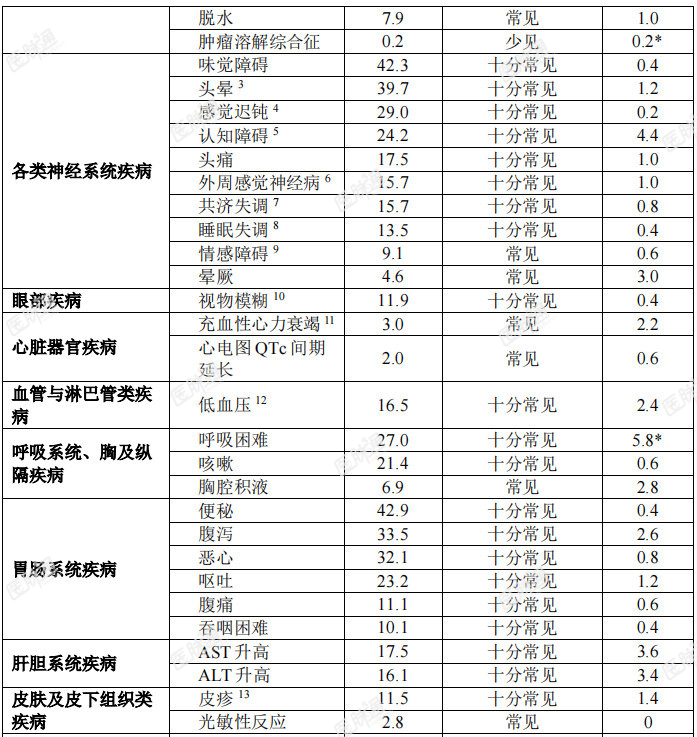
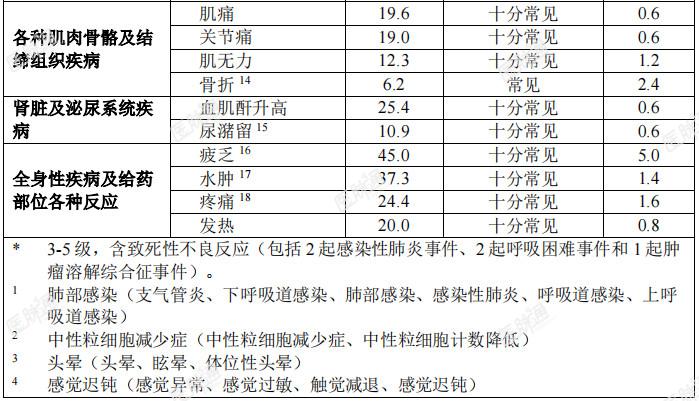
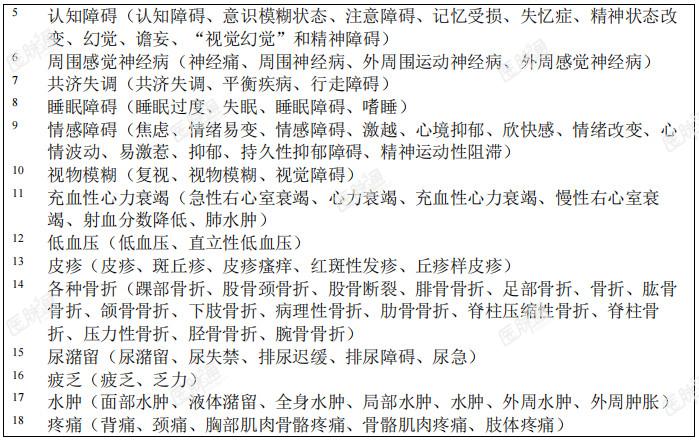
表6:在临床试验中接受恩曲替尼治疗的儿童患者发生的药物不良反应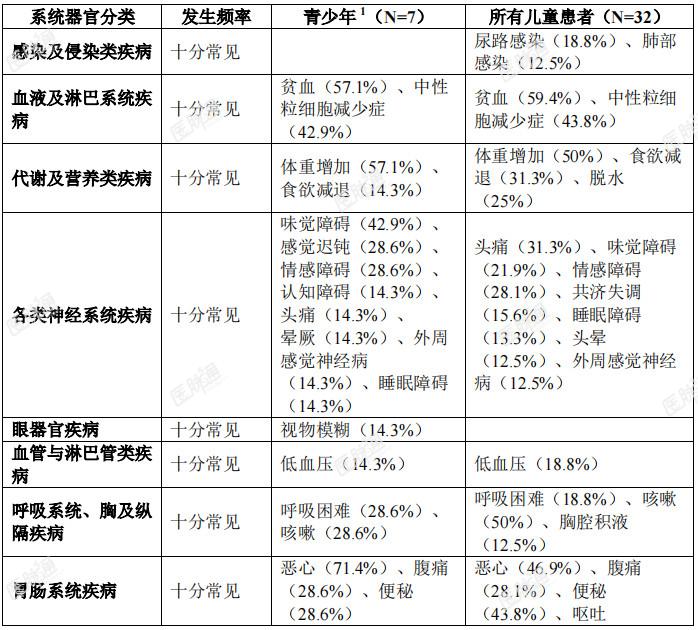
特定药物不良反应描述:认知障碍:临床试验中曾报告了各种认知受损症状。此类症状包括认知障碍(6.3%)、意识模糊状态(7.3%)、注意障碍(3.8%)、记忆受损(4.2%)、失忆症(2.8%)、精神状态改变(1.2%)、幻觉(1.0%)、谵妄(0.8%)、视觉幻觉(0.4%)和精神障碍(0.2%)。有4.4%的患者报告3级事件。儿童人群中有3.4%(1/29 例)发生1级注意障碍。基线时有脑转移的患者其发生率(29.7%)高于无脑转移者(23.1%)。发生认知障碍的中位起始时间为0.92个月。骨折:分别有5.3%(25/475)的成人患者和21.9%(7/32)的儿童患者发生骨折。总体而言,对骨折部位肿瘤累及的评估不充分;但有些患者的影像学异常可能提示肿瘤对骨的浸润。成人和儿童患者的大多数骨折发生在髋关节或下肢其他部位(例如股骨或胫骨干)。有2例儿童患者发生双侧股骨颈骨折。未见因骨折停止恩曲替尼治疗的患者。成人患者的某些骨折发生在跌倒或疾病累及部位遭受其他外伤的情况下。成人患者发生骨折的中位时间为3.42个月(范围:0.26-18.5个月)。发生骨折的成人患者中,有36.0%因骨折暂停恩曲替尼治疗。儿童患者的所有骨折均发生在遭受极轻微外伤或无外伤的患者中。7例儿童患者共报告11例骨折不良反应。儿童患者发生骨折的中位时间为4.3个月(范围:2.46-7.39个月)。发生骨折的儿童患者中,有42.9%(3/7)暂停恩曲替尼治疗。3例骨折为2级,4例骨折为3级。3例3级骨折为严重事件。无骨折部位肿瘤受累报告。除一例骨折事件外,所有骨折均已恢复。共济失调:有15.7%的患者报告共济失调(包括共济失调、平衡障碍和步态障碍等事件)。发生共济失调的中位时间为0.4个月(范围:0.03-28.19个月),中位持续时间为0.7个月(范围:0.03-11.99个月)。大多数患者(67.1%)的共济失调可恢复。老年患者(23.8%)的共济失调相关不良事件发生率高于年龄<65岁者(12.8%)。晕厥:有4.6%的患者报告晕厥事件。在部分患者中,晕厥伴随低血压、脱水或QTc间期延长的报告。QTc间期延长:在临床试验中接受恩曲替尼治疗的504例患者中,进行过至少一次基线后ECG评估的患者中有17例(4.0%)在开始恩曲替尼治疗后发生QTcF间期延长大于60ms,12例(2.8%)QTcF间期超过500ms。周围感觉神经病:有15.7%的患者报告周围感觉神经病。中位发生时间为0.49个月(范围:0.03-20.93个月),中位持续时间为0.8个月(范围:0.07-6.01个月)。大多数患者(55.7%)的周围神经病可痊愈。眼部疾病:临床试验中报告的眼部疾病包括视物模糊(8.5%)、复视(2.6%)和视觉障碍(1.6%)等事件。发生眼器官疾病的中位时间为1.9个月(范围:0.03-21.59个月)。眼部疾病的中位持续时间为1个月(范围:0.03-14.49个月)。大多数患者(61.7%)的眼部疾病事件可痊愈。儿童人群:儿童人群的总体安全性特征与成人用药的安全性特征相似。儿童患者的安全性是根据3项携带NTRK融合基因的成人实体瘤患者的开放、单臂临床试验(ALKA、STARTRK-1和STARTRK-2)所得数据以及32例儿童患者数据(30例入组STARTRK-NG的患者及2例入组STARTRK-2的患者)的外推结果确立的。其中2例患者未满2岁,23例患者介于2-11 岁,7例患者介于12-17岁。儿童患者中发生频率高于(发生率至少高 5%)成人患者的3级或4级不良反应和实验室检测异常分别为中性粒细胞减少症(28.1%vs.3.4%)、体重增加(21.9%vs.6.9%)、头痛(6.3%vs.0.6%)和骨折(12.5%vs.1.9%)。青少年中的安全性数据有限,但青少年中的安全性特征与本品的总体安全性特征相似。青少年中报告的≥ 3级不良反应为中性粒细胞减少症和头痛。老年患者:在临床试验中接受治疗的504例患者中,130例(25.8%)患者年满65岁,34例(6.7%)年满75岁。恩曲替尼用于老年患者的总体安全性特征与小于65岁的患者中的安全性特征相似。老年患者中的发生频率高于小于65岁患者的不良反应为头晕(48.5%vs36.6%)、血肌酐升高(31.5%vs23.3%)、低血压(21.5%vs14.7%)、共济失调(23.8%vs12.8%)。上市后经验:不适用。
一般事项:不同肿瘤类型的有效性:恩曲替尼的治疗获益是基于单臂研究确立的,研究入选的是样本量相对较小的一群携带NTRK融合基因的实体瘤患者。在有限的几种实体瘤类型中,总体缓解率和缓解持续时间均证明了恩曲替尼具有良好的疗效。该疗效可能会因为患者的肿瘤类型以及是否合并其它基因突变而存在程度上的差异。鉴于此,患者仅在无满意治疗选择时(如:可选择的治疗手段的临床获益尚未明确,或该治疗手段已被使用),才选择恩曲替尼。认知障碍:恩曲替尼临床试验中曾有过认知障碍的报告,包括意识模糊、精神状态改变、记忆障碍和幻觉。年龄大于65岁的患者中的这些事件发生率高于较年轻患者。应监测患者是否出现认知改变的体征。应根据认知障碍的严重程度,按用法用量表4所述调整恩曲替尼治疗。
应告知患者,恩曲替尼治疗有可能引起认知改变。应指导患者,如果患者出现认知障碍症状,在症状消退之前,应避免驾驶或操纵机器。(参见注意事项中“驾驶和操纵机器的能力”)。骨折:恩曲替尼可导致骨折风险增加(参见不良反应中“特定药物不良反应描述”)。应及时评价患者的骨折症状或体征(例如疼痛、运动变化、畸形)。成人患者的某些骨折发生在跌倒或疾病累及部位遭受其他外伤的情况下,儿童患者的骨折均发生在遭受极轻微外伤或无外伤的患者中。关于恩曲替尼是否影响现有骨折的愈合或今后发生骨折风险,尚无数据。大多数儿童患者继续接受恩曲替尼治疗,且骨折痊愈。高尿酸血症:在恩曲替尼治疗患者中曾观测到高尿酸血症。在开始恩曲替尼治疗前和治疗期间应定期评估血清尿酸水平。应监测患者是否出现高尿酸血症体征和症状。应根据临床指征开始降尿酸治疗,并在观察到高尿酸血症症状和体征时暂停恩曲替尼治疗。应根据严重程度,按用法用量表4所述调整恩曲替尼治疗。充血性心力衰竭:恩曲替尼各项临床试验中均有充血性心力衰竭(CHF)的报告(参见不良反应中的表5)。在既往有或无心脏病史的患者中均有观察到这些反应,在给予利尿剂和/或减量/中断恩曲替尼的治疗后不良反应痊愈。对于出现CHF症状或有已知风险因素的患者,应在开始恩曲替尼治疗前评估左心室射血分数(LVEF),并在用药过程中密切监测,对有CHF临床体征和症状(包括呼吸短促或水肿)的患者进行评估,并给予适当的临床治疗。应根据CHF严重程度,按用法用量表4所述调整恩曲替尼治疗。QTc间期延长:临床试验中,在恩曲替尼治疗的患者中观察到QT间期延长。基线QTc间期大于450ms的患者、先天性长QT综合征的患者和服用已知会延长QT间期的药物的患者应避免使用恩曲替尼。电解质失衡或重大心脏疾病,包括近期心肌梗死、充血性心力衰竭、不稳定型心绞痛和缓慢型心律失常患者应避免使用恩曲替尼。如果治疗医师认为恩曲替尼对存在上述任何病症的患者的潜在获益大于潜在风险,则应进行额外监测,并考虑专科医师会诊。建议在基线时及恩曲替尼治疗1个月后进行心电图及电解质评估,并在整个恩曲替尼治疗期间根据临床指征定期监测心电图和电解质。应根据 QTc 间期延长严重程度,按用法用量表4所述调整恩曲替尼治疗。有生育能力的女性:孕妇使用恩曲替尼可能会对胎儿造成伤害。有生育能力的女性患者必须在恩曲替尼治疗期间和末次给药后5周内采取高效避孕措施。女性伴侣有生育能力的男性患者必须在恩曲替尼治疗期间以及末次给药后3个月内采用高效避孕方法。乳糖不耐受:本品含有乳糖。存在半乳糖不耐受、乳糖酶完全缺乏或葡萄糖-半乳糖吸收不良等罕见的先天性疾病的患者不应使用本品。日落黄:本品200mg硬胶囊含有辅料日落黄,可能会引起过敏反应。药物滥用和药物依赖性:不适用。驾驶和操纵机器的能力:恩曲替尼可能影响驾驶和操纵器械的能力。应告知患者,在恩曲替尼治疗期间出现认知不良反应、晕厥、视物模糊或头晕时避免驾驶或操纵机械,直至症状消退。
恩曲替尼临床试验尚无用药过量经验。应密切监测用药过量患者,并给予支持性治疗。尚无已知的恩曲替尼解毒剂。
具有生育能力的女性和男性:生育力:尚未在动物中开展评价恩曲替尼影响的生育力研究。妊娠检查:有生育能力的女性患者在开始恩曲替尼治疗前应在有医疗人员监督的情况下进行妊娠检测。避孕:有生育能力的女性患者必须在恩曲替尼治疗期间和末次给药后5周内采取高效避孕措施。目前尚不清楚恩曲替尼是否会降低全身作用的激素避孕药的有效性。因此,应建议使用全身作用的激素避孕药的女性增加屏障避孕法。基于潜在遗传毒性,有具备生育能力的女性伴侣的男性患者必须在恩曲替尼治疗期间以及末次给药后3个月内采用高效避孕方法。妊娠:必须告知育龄期女性患者在接受恩曲替尼治疗期间避免妊娠。尚未获得恩曲替尼用于孕妇的数据。根据恩曲替尼动物研究结果及其作用机制,孕妇服用恩曲替尼可能会导致胎儿损害。应向接受恩曲替尼治疗的患者告知本品对胎儿的潜在危害。应告知女性患者在怀孕后联系医生。分娩:目前尚未确立恩曲替尼在生产与分娩时使用的安全性。哺乳:尚不清楚恩曲替尼或其代谢产物是否会分泌至人乳汁中。未开展旨在评估恩曲替尼对乳汁分泌的影响或乳汁中是否含恩曲替尼的研究。因为恩曲替尼对哺乳期婴儿的潜在危害不详,因此建议母亲在恩曲替尼用药期间停止哺乳。实体瘤:本品用于年满12岁以上儿童患者的有效性是根据3项携带 NTRK 融合基因实体瘤成人患者的开放、单臂临床试验(ALKA、STARTRK-1和STARTRK-2)所得数据以及入组STARTRK-NG的青少年患者的药代动力学数据外推确立的。年满12岁的儿童患者给予本品剂量(基于体表面积)后产生的全身暴露量与成人接受600mg本品后的暴露量相似。相比成人患者,恩曲替尼的使用在儿童患者中与更高的骨折发生率相关。本品用于未满12岁携带NTRK融合基因实体瘤儿童患者的安全性和有效性尚未确立。非小细胞肺癌(NSCLC):本品用于ROS1阳性局部晚期或转移性非小细胞肺癌(NSCLC)儿童患者的安全性和有效性尚未确立。年龄≥65岁的患者和较年轻的患者未观察到安全性或有效性的差异。年龄≥65岁的患者无需调整剂量。“特殊人群剂量说明”
恩曲替尼对其他药物的影响:CYP底物:恩曲替尼是CYP3A4的弱抑制剂。恩曲替尼单次口服给药与口服咪达唑仑(一种敏感性CYP3A底物)合并导致患者的咪达唑仑AUC升高50%,但导致咪达唑仑Cmax下降21%。由于药物不良反应风险增加,恩曲替尼与治疗范围较窄的敏感性CYP3A4底物(例如西沙必利、环孢菌素、麦角胺、芬太尼、匹莫齐特、奎尼丁、他克莫司、阿芬太尼和西罗莫司)合并给药时应谨慎。P-gp底物:体外数据表明,恩曲替尼对P-gp有潜在的抑制作用。恩曲替尼单次口服给药与地高辛(一种敏感性P-gp底物)合并导致地高辛的Cmax升高约28%,总体暴露量(AUC)升高约18%。地高辛单独给药和地高辛+恩曲替尼合并给药的地高辛肾脏清除率相似,提示恩曲替尼对地高辛肾脏清除率的影响极低。认为恩曲替尼对地高辛吸收的影响无临床意义,但尚不清楚恩曲替尼对更敏感的口服P-gp底物(如达比加群酯)的影响是否可能更大。BCRP底物:与P-gp结果相似,在体外研究中观察到对BCRP有轻度抑制作用。这种抑制作用的临床意义尚不清楚,但由于存在吸收增加的风险,敏感的口服BCRP底物(例如甲氨蝶呤、米托蒽醌、托泊替康、拉帕替尼)与恩曲替尼合并给药时应谨慎。
其他转运蛋白底物:体外数据表明,恩曲替尼对有机阴离子转运多肽(OATP)1B1有较弱的潜在抑制作用。这种抑制作用的临床意义尚不清楚,但由于存在吸收增加的风险,敏感的口服OATP1B1底物(例如阿托伐他汀、普伐他汀、瑞舒伐他汀、瑞格列奈、波生坦)与恩曲替尼合并给药时应谨慎。PXR调节的酶底物:体外研究表明,恩曲替尼可能会诱导孕烷X受体(PXR)调节的酶(例如CYP2C家族和UGT)。恩曲替尼与 CYP2C8、CYP2C9或CYP2C19底物(例如瑞格列奈、华法林、甲苯磺丁脲或奥美拉唑)合并给药可降低其暴露量。口服避孕药:
目前尚不清楚恩曲替尼是否会降低全身作用的激素避孕药的有效性。因此,建议使用全身作用的激素避孕药的女性增加屏障避孕法。其他药物对恩曲替尼的影响:基于体外数据,CYP3A4是介导恩曲替尼代谢和生成主要活性代谢产物M5的主要酶。CYP3A或p-gp诱导剂:利福平(一种强效CYP3A诱导剂)多次口服给药与恩曲替尼单次口服给药合并时,恩曲替尼的全身暴露量下降77%,Cmax下降56%。应避免恩曲替尼与CYP3A诱导剂(包括但不限于卡马西平、苯巴比妥、苯妥英、利福布汀、利福平和圣约翰草[贯叶连翘]、阿帕他胺、利托那韦)合并用药。CYP3A或P-gp抑制剂:恩曲替尼单次口服给药与伊曲康唑(一种强效CYP3A4抑制剂)多次口服给药合并时,AUCinf升高600%,Cmax升高173%。避免强效和中效CYP3A抑制剂(包括但不限于利托那韦、沙奎那韦、酮康唑、伊曲康唑、伏立康唑、泊沙康唑、葡萄柚或塞维利亚柑橘)与恩曲替尼合并用药。如无法避免合并用药,需按用法用量所述调整恩曲替尼剂量。尽管抑制性P-gp药品预计对恩曲替尼药代动力学无明显影响,但由于存在恩曲替尼暴露量增加的风险,强效或中效P-gp抑制剂(例如维拉帕米、硝苯地平、非洛地平、氟伏沙明、帕罗西汀)与恩曲替尼合并用药时应谨慎。致胃液pH值升高的药品:恩曲替尼的体外水溶性具有pH值依赖性。临床研究显示,恩曲替尼与兰索拉唑(质子泵抑制剂[PPI])合并用药导致恩曲替尼全身暴露量下降25%,Cmax下降23%,但无临床意义。因此,恩曲替尼与PPI或能导致胃液pH值升高的其他药物(例如H2受体拮抗剂或抗酸剂)合并用药时,无需调整剂量。
Mayne Pharma Inc.

86981735000070;86981735000087
NTRK融合阳性实体瘤:将三项单臂、开放性临床试验(ALKA、STARTRK-1和STARTRK-2)的NTRK融合阳性实体瘤成人患者进行合并,通过预先规定的整合分析计划,评价了恩曲替尼治疗NTRK融合基因阳性实体瘤成人患者的有效性。研究ALKA是一项针对肿瘤携带NTRK1/2/3、ROS1或ALK分子变异、年龄≥18岁的实体瘤患者开展的一项I期、单臂、开放研究,目的是确定最大耐受剂量。研究STARTRK-1是一项针对肿瘤携带NTRK1/2/3、ROS1或ALK分子变异、年龄≥18岁的实体瘤患者开展的一项I期、多中心、单臂、开放研究。该研究包含一个剂量递增阶段和一个剂量扩展阶段。在剂量扩展阶段,患者接受恩曲替尼600mg每日一次的治疗,每4周为一疗程,主要目的是评价II期推荐剂量。研究STARTRK-2是针对肿瘤携带NTRK1/2/3、ROS1或ALK基因重排的实体瘤患者开展的一项多中心、国际性、单臂、篮式II期研究。患者接受恩曲替尼600mg每日给药一次,每4周为一疗程。整合分析的主要有效性终点为盲态独立中心审查(BICR)委员会根据实体瘤疗效评价标准RECISTv1.1评估的客观缓解率(ORR)和缓解持续时间(DOR)。对于基线时存在中枢神经系统(CNS)转移的患者将评估颅内客观缓解率(IC-ORR)、颅内缓解持续时间(IC-DOR)及颅内无进展生存期(ICPFS)(均由BICR根据RECISTv1.1进行评估)。整合的有效性可评价人群共包含74例经确证为NTRK融合阳性、接受恩曲替尼治疗的实体瘤成人患者,这些患者既往未接受过TRK抑制剂治疗,基线时经研究者评估存在可测量的病灶,且随访时间≥6个月。在入组研究前,由《临床实验室改进修正案》(CLIA)认证的或同等资质认证的实验室采用经验证的基于核酸的检测方法确证NTRK融合阳性状态。有效性可评价人群的基线人口统计学和疾病特征为:47.3%男性,中位年龄57岁(范围:21-83岁),70.0%为白人(高加索人种),17.6%为亚洲人,5.5%为西班牙裔或拉丁美洲裔,59.7%从不吸烟。基线ECOG(东部肿瘤协作组)体能状态评分为0(40.5%)、1(45.9%)或2(13.5%)。大多数患者(97.3%)有转移病灶,其中最常见的转移部位为肺部(60.8%)、淋巴结(52.7%)和脑部(25.7%),2.7%的患者肿瘤为局晚期,27%的患者既往未接受全身治疗。总体中位随访时间为14.2个月。NTRK融合阳性实体瘤患者的有效性结果总结参见表7。 表7:NTRK 融合阳性实体瘤成人患者中由 BICR 评估的总体有效性总结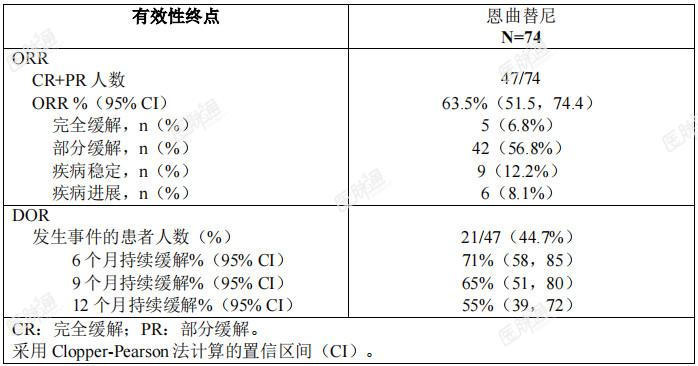
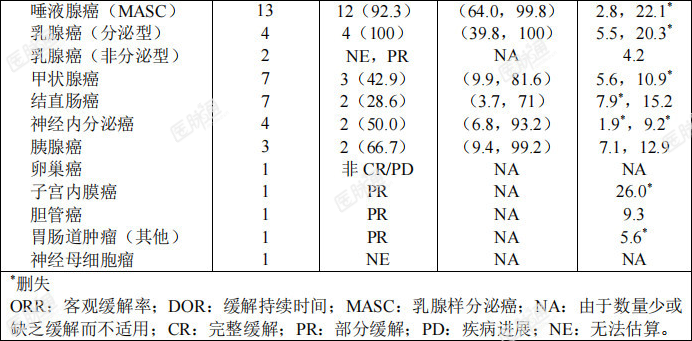
所有有效性可评价的NTRK融合阳性实体瘤成人患者中按肿瘤类型总结的客观缓解率和缓解持续时间参见表8。表8:NTRK融合阳性实体瘤成人患者中按肿瘤类型总结的有效性总结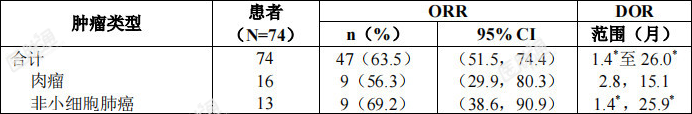
颅内缓解:在74例NTRK融合阳性实体瘤患者的整合有效性可评价人群中,16例患者经BICR评估基线时存在CNS转移,其中8例CNS病灶可测量。由BICR根据RECISTv1.1评估,这8例患者中有5例达到颅内(IC)缓解(1例CR和4例PR),ORR为62.5%(95% CI:24.5,91.5),DOR为NE(5.0,NE)。在开始本品治疗前2个月内,这8例患者中有4例接受了颅内放疗。原发性CNS肿瘤:在这三项试验中,7例原发性CNS肿瘤的成人患者接受了恩曲替尼治疗,并进行了至少6个月随访。由BICR根据神经肿瘤学疗效评估标准(RANO)评估,一例患者达到客观缓解。
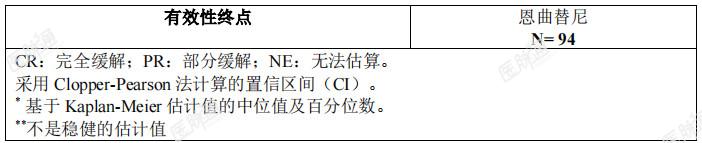
本适应症为基于替代终点获得附条件批准上市,暂未获得临床终点数据,有效性和安全性尚待上市后进一步确证。ROS1阳性非小细胞肺癌(NSCLC):将三项单臂、开放性临床试验(ALKA、STARTRK-1和STARTRK-2)中ROS1阳性局部晚期或转移性NSCLC患者进行合并,通过预先规定的整合分析计划,评价了恩曲替尼治疗ROS1阳性局部晚期或转移性NSCLC患者的有效性。整合分析中的主要有效性终点为盲态独立中心审查(BICR)委员会根据实体瘤疗效评价标准 RECISTv1.1评估的客观缓解率(ORR)和缓解持续时间(DOR)。次要有效性终点包括无进展生存期(PFS)、至中枢神经系统(CNS)进展时间、总生存期(OS)、基线时存在中枢神经系统(CNS)转移的患者的颅内客观缓解率(IC-ORR)与颅内缓解持续时间(IC-DOR)及颅内无进展生存期(IC-PFS)(均由BICR根据RECISTv1.1评价)。有效性可评价分析人群共包含94例经组织学确证为ROS1阳性、接受恩曲替尼治疗的NSCLC患者,这些患者既往未接受过ROS1抑制剂治疗,基线时经研究者评估存在可测量的病灶,且随访时间≥12个月。在入组研究前,由CLIA认证的或同等资质认证的实验室采用经验证的基于核酸的检测方法确证ROS1阳性状态。有效性可评价人群的基线人口统计学和疾病特征为:36.2%男性,中位年龄53岁(范围:27-86岁),48.9%为白人(高加索人种),43.6%为亚洲人,5.3%为黑人,2.4%为西班牙裔或拉丁美洲裔,59.6%从不吸烟。基线ECOG(东部肿瘤协作组)体能状态评分为0(37.2%)、1(51.1%)或2(11.7%)。大多数患者(98.9%)有转移病灶,其中42.6%为脑转移(其他常见转移部位为肺部[57.4%]和淋巴结[75.5%]),1.1%的患者肿瘤为局部晚期,且33%的患者既往未接受全身治疗。中位随访持续时间为20.9个月。ROS1阳性NSCLC患者的有效性结果总结参见表9。 表9:ROS1阳性NSCLC患者中由BICR评估的总体有效性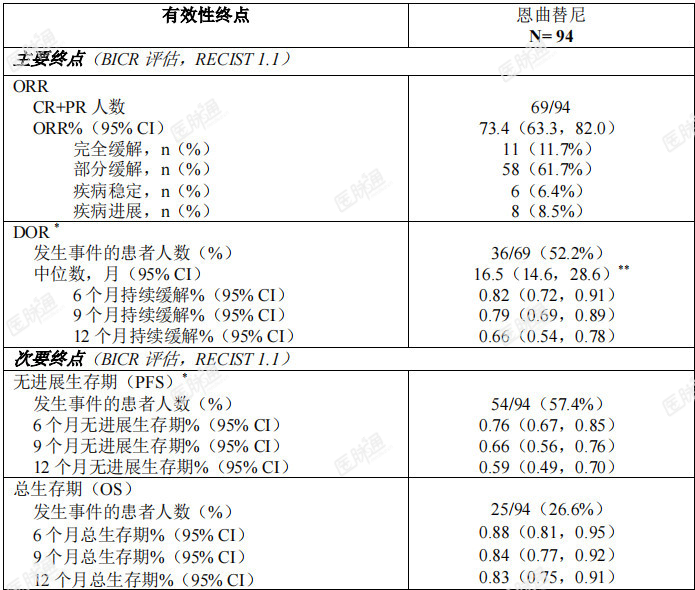
颅内缓解:在94例ROS1阳性NSCLC患者的有效性可评价分析人群中,34例患者经BICR评估基线时存在CNS转移,其中18例CNS病灶可测量。这18例患者中有14例达到颅内(IC)缓解(2例CR和12例PR),ORR为77.8%(95% CI:52.4,93.6),DOR≥6个月、≥9个月及≥12个月的患者百分比(95%CI)分别为68%(41%,94%)、59%(31%,87%)和51%(22%,79%)(Kaplan-Meier估计值)。在开始本品治疗前2个月内,这18例患者中有8例接受了颅内放疗。
胶囊剂
200mg*90粒
口服固体药用高密度聚乙烯瓶及口服固体药用防潮组合瓶盖包装。 200mg胶囊:90粒/瓶,1瓶/盒 100mg胶囊:30粒/瓶,1瓶/盒
密封,不超过30℃保存。
36个月。
国药准字 HJ20220068;国药准字 HJ20220069 附条件批准上市

微信扫一扫,关注医库最新动态