克尔来福/CoronaVac说明书
COVID-19 Vaccine (Vero Cell), Inactivated
灭活的新型冠状病毒19nCoV-CDC-Tan-HB02株
本品系用新型冠状病毒(CZ02 株)接种非洲绿猴肾细胞(简称 Vero 细胞),经培养、收获病毒液、灭活病毒、浓缩、纯化和氢氧化铝吸附制成。本品不含防腐剂。
主要成分:灭活的新型冠状病毒(CZ02 株)。
佐剂:氢氧化铝佐剂。
辅料:磷酸氢二钠十二水合物、磷酸二氢钠一水合物、氯化钠。
本品为乳白色混悬液,可因沉淀而分层,易摇散。
作用与用途本品适用于预防新型冠状病毒(SARS-COV-2)感染所致的疾病(COVID--19)。
本品为基于境外 III 期临床保护效力试验两个月的结果获得附条件批准上市,暂未获得最终分析数据,有效性和安全性结果尚待进一步最终确证。
免疫程序和剂量
本品基础免疫为 2 剂次,间隔 14~28 天;每一次人用剂量为 0.5 ml。
推荐的接种途径为肌肉注射,最佳部位为上臂三角肌,注射前须摇匀。
尚未确定本品是否需要进行加强免疫。
接种对象
本品适用于 18 岁及以上人群的预防接种。
本品境外期临床试验中 60 岁及以上人群所占比例较低(5.10%),60 岁及以上人群保护效力证据尚不充分后续临床试验将继续开展 60 岁及以上人群保护效力研究,进一步获取该人群保护效力证据。已有的临床试验数据显示,60 岁及以上人群接种本品后产生一定程度的中和抗体。疾病预防控制相关机构接种使用时,需结合 60 岁及以上人群健康状态和暴露风险,评估接种本品的必要性。
在境内外开展的两项临床试验中评价本品的安全性。第-项为境内随机、双盲、安慰剂平 行对照的1 1 II 期临床试验,初步评价本品在18岁及以上人群中的安全性和免疫原性。第 二项为国际多中心、随机、双盲、安慰剂平行对照的I期临床试验,评价本品的保护效力、 安全性和免疫原性。研究者主动随访每剂接种后0-21/28天的安全性数据,观察不良事件 发生情况,同时关注全程接种后12个月内发生的严重不良事件。 1.本品临床试验不良反应发生情况总述 按照国际医学科学组织委员会( CIOMS )推荐的不良反应发生率分类:十分常见(≥10% ), 常见( 1-10%,含1%),偶见(0.1-1%,含0.1%),罕见(0.01-0.1%,含0.01%), 十分罕见(<0.01%),汇总本品I1II期和I期临床试验研究人群安全性数据进行如下描述: (1 )接种部位不良反应 十分常见:疼痛; 偶见:红晕、肿胀、硬结、皮疹、瘙痒; 罕见:红斑。 (2)全身不良反应 十分常见:头痛; 常见:发热、疲劳/乏力、肌肉痛、关节痛、咳嗽、呼吸困难、恶心、腹泻、皮肤瘙痒; 偶见:头晕、厌食、呕吐、口咽疼痛、吞咽困难、流涕、便秘、超敏反应; 罕见:急性过敏反应、嗜睡、困倦、入睡困难、喷嚏、鼻咽炎、鼻充血、咽干、流行性感冒、 感觉减退、肢体疼痛、心悸、腹痛、皮疹、皮肤黏膜异常、痤疮、眼痛、耳部不适、淋巴结病; 十分罕见:寒颤、味觉障碍、味觉丧失、感觉异常、震颤、注意障碍、鼻衄、哮喘、咽喉刺激、 扁桃体炎、肢体不适、颈部疼痛、颌骨疼痛、颈部肿块、口腔溃疡、牙疼、食管疾病、胃炎、 粪便变色、眼痛、视物模糊、眼刺激、视力减退、耳痛、紧张、高血压、低血压、尿失禁、 月经延迟。 (3)不良反应严重程度 本品临床试验中观察到的不良反应严重程度以1级(轻度)为主,3级及以上征集性不良 反应发生率为0.44%,未报告与接种本品相关的4级不良反应。临床试验报告的3级接种 部位不良反应为疼痛、皮疹、瘙痒; 3级全身不良反应为发热、疲劳1乏力、头痛、肌肉痛、 关节痛、咳嗽、呼吸困难、呕吐、腹泻、便秘、吞咽困难。 (4)严重不良事件( SAE ) 截至2020年10月31日,境外I期临床试验观察到的严重不良事件中,有1例受试者接 种本品后出现较为严重的恶心、呕吐等症状,入院经药物治疗后已痊愈,研究者判断与接 种本品有关。另有1例受试者接种本品后出现“右上肢无力、口齿不清”症状,就医后当 地医院以“炎性脱髓鞘综合症;多发性硬化症(MS) ;临床孤立综合征(CIS) ;急性播 散性脑脊髓炎( ADEM)待排除”收入院。目前尚无法确定该病例与接种本品的相关性。 2.本品境内外各项临床试验不良反应具体发生情况 (1)境内1/II期临床试验 境内I / II 期临床试验共入组1120名18岁及以上受试者,其中420例受试者至少接种1 剂本品( I / II 期临床试验中剂量组),男性192名( 45.71%),女性228人( 54.29%)。 截至2020年10月13日,已完成全程免后至少28天的安全性随访,长期安全性随访尚在 进行中。 (2)境外II期临床试验 境外II期临床试验计划入组45000名18岁及以上受试者。截至2020年10月31日期中分 析时,14312 例受试者至少接种1剂本品。其中,男性12088人( 84.46% ),女性2224人(15.54%);18~59岁14209人(99.28%),60岁及以上103人(0.72%)。已完成全程免后至少28天的安全性随访,长期安全性随访尚在进行中。尚未发现60岁及以上人群安全性风险信号的增加。II 期临床试验中观察到的非征集性不良反应发生率为16.03%,3级及以上发生率为0.23%。非征集性不良反应中较征集性不良反应新增严重程度为3级的症状包括:口咽痛(0.01%)、非接种部位皮疹(0.01%)、淋巴结病(0.01%)、超敏反应(0.01% )。
1、对本品中的活性成分、任何一种非活性成分生产工艺中使用的物质过敏者,或以前接种本品或同类疫苗时出现过敏者。
2、既往发生过疫苗严重过敏反应者(如急性过敏反应、血管神经性水肿、呼吸困难等)。
3、患有严重神经系统疾病者(如横贯性脊髓炎、格林巴利综合症、脱髓鞘疾病等)。
4、未控制的严重慢性病患者。
5、妊娠期及哺乳期妇女。
1、目前暂未获得本品的保护持久性数据, 接种后仍需根据疫情防控需要采取必要的防护措施。
2、目前本品对 60 岁及以上人群的保护效力数据有限, 疾病预防控制相关机构接种使用时, 需结合该人群健康状态和暴露风险,评估接种本品的必要性。
3、本品严禁血管内注射。尚无本品采用皮下或皮内注射的安全性和有效性数据。
4、使用前应检查包装容器、标签、外观、有效期是否符合要求,如玻璃针管有裂纹,玻璃针管外表面有斑点污点、擦痕,标签不清或超过有效期时限及外观异常等均不得使用。
5、开启疫苗瓶和注射时,切勿使消毒剂接触疫苗。
6、本品须置于儿童不可触及处。
7、应备有肾上腺素等药物和设备,以备发生严重急性过敏反应时急救用。在接种本品后应在现场观察至少 30 分钟。
8、本品不能与其他疫苗在同一注射器内混合。
9、本品严禁冻结。开启后应马上使用。
10、患急性疾病慢性疾病的急性发作期、严重慢性疾病过敏体质和发热者需慎用;必要时经医生评估后延迟接种。
12、患有血小板减少症或者出血性疾病者,肌肉注射本品可能会引起出血,需慎用。
13、尚未获得本品对免疫功能受损者(例如恶性肿瘤、肾病综合征、艾滋病患者)的安全性和有效性数据,此类人群接种本品应基于个体化考虑。
14、注射人免疫球蛋白者应至少间隔 1 个月以上接种本品,以免影响免疫效果。
15、尚未进行同期(先、后或同时)接种其它疫苗对本品免疫原性影响的临床研究,同期接种其它疫苗时应咨询专业医师。
16、接种本品后出现任何神经系统不良反应者,禁止再次使用。
17、与其它疫苗一样,无法确保本品对所有接种者均产生保护作用。
育龄期妇女:在临床试验中接种本品后意外妊娠的妇女中收集到的数据非常有限,尚不足以判断接种本品后可能导致发生不良妊娠结局的风险。妊娠期或哺乳期女性:目前尚未获得孕妇及哺乳期妇女使用本品的临床试验数据。目前本品对 60 岁及以上人群的保护效力数据有限, 疾病预防控制相关机构接种使用时, 需结合该人群健康状态和暴露风险,评估接种本品的必要性。
北京科兴生物制品有限公司;北京科兴中维生物技术有限公司
86983009000038,86983009000021
1.保护效力试验结果
本品关键性Ⅲ期临床试验采用多中心、随机、双盲、安慰剂平行对照的设计,分别在巴西 18 及以上医务人员和土耳其 18~59 岁健康人群中开展,用于评估本品在高危人群(接诊 COVID-19 患者的医务人员)和普通人群中的保护效力。
主要研究假设为:在上述人群中按照 0,14 天程序接种 2 剂本品 14 天后,相较于安慰剂组的保护效力(VE)95% 置信区间下限大于 30%。巴西疫苗保护效力的主要分析方法基于人年发病率计算保护率,土耳其疫苗保护效力则基于发病率计算保护率。所有有效终点病例均经过终点判定委员会确认。
*(1)巴西期临床试验 *
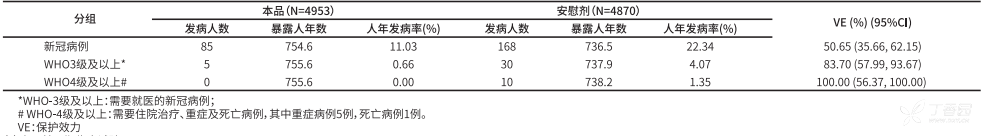
巴西 III 期临床试验目标人群为接诊 COVID-19 患者的医务人员,试验共入组 12,396 名受试者,获得 253 例监测期有效病例。按照 0,14 天程序接种 2 剂本品 14 天后预防由型冠状病毒所致疾病(COVID--19)的保护效力:对住院、重症及死亡新冠病例的保护效力为 100.00%(95%CI:56.37-100.00),对有明显症状且需要医的新冠病例保护效力为 83.70%(95%CI:57.99-93.67),对包括轻微症状不需就医的所有新冠病例保护效为 50.65%(95%CI:35.66-62.15)。接种本品受试者平均随访时间为 70.3±25.6 天,随访中位时间为 73.0 天。
*表 3.巴西 III 期临床试验 2 剂免疫 14 天后对 COVID--19 的保护效果(ppS) *
(2)土耳其期临床试验
土耳其期临床试验目标人群为处于高风险的医护人员(K-1)和处于正常风险的一般人群(K-2),截止 2020 年 12 月 23 日,共完成 K-1 队列受试者入组 918 例,K-2 队列入组受试者 6453 例,总计 7371 例;其中 1322 例受试者完成两剂接种并进入第二剂接种后 14 天观测期。基于 29 例病例的分析结果显示,按照 0,14 天程序接种 2 剂本品 14 天后预防 COVID--19 的保护效力为 91.25%(95%C:71.25-97.34),结果详见表 4。
*表 4.土耳其期临床试验 2 剂免疫 14 天后对 COVD-19 的保护效果 *

2.免疫原性
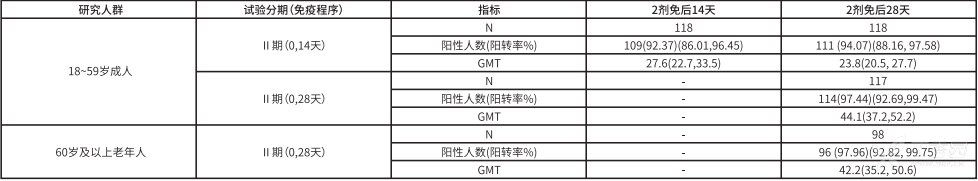
本品免疫原性终点包括血清中和抗体阳转率和几何平均滴度(GMT)阳转定义为免疫前中和抗体滴度<1:8 者,免疫后中和抗体滴度 ≥ 1:8;或免疫前中和抗体滴度 ≥ 1:8 者,免疫后中和抗体滴度达 4 倍及以上增长。中和抗体测定采用细胞培养微量中和试验(细胞病变法)确定。
*表 5.18 岁及以上人群不同免疫程序中和抗体阳转率和 GMT(95%CI)(PPS)。 *
3.交叉中和
基于 80 名 26-45 岁受试者按照 0,14 天程序免疫前后的血清对境内外流行的 12 种新冠病毒株(CZ02、WZL、WGF、ZJY、SSH、JWL、ZYF、HAC、HJL、ZXZ、QHF 和 NOOR)进行血清交叉中和检测。采用细胞培养微量中和试验(细胞病变法)进行血清中和抗体检测结果显示,本品免疫后诱导机体产生的抗体具有交叉中和不同新冠病毒株(含 D614 G 突变株)的能力,阳转率在 80.00%-100.00% 之间,GMT 在 15.4-46.7 之间。
注射剂
0.5ml/支,每1次人用剂量为0.5ml,含灭活新型冠状病毒抗原600SU;1.0ml/瓶(2次人用剂量),含灭活新型冠状病毒抗原1200SU。每1次人用剂量为0.5ml,含灭活新型冠状病毒抗原600SU;
国药准字S20210003,国药准字S20210002

微信扫一扫,关注医库最新动态