众爱可维说明书
COVID-19 Vaccine (Vero Cell), Inactivated
灭活的新型冠状病毒19nCoV-CDC-Tan-HB02株
本品系用新型冠状病毒 WIV04 株,接种非洲绿猴肾细胞(Vero 细胞),经培养、收获、灭活、澄清、浓缩、纯化,经氢氧化铝佐剂吸附后制成。不含抗生素和防腐剂。
有效成分:灭活的新型冠状病毒 WIV04 株。
佐剂:氢氧化铝佐剂。
辅料:氯化钠、磷酸氢二钠、磷酸二氢钠。
本品为乳白色混悬液,可因沉淀而分层,易摇散。
本品适用于预防由新型冠状病毒(SARS-CoV-2)感染引起的疾病(COVID--19)。本品基于境外期临床保护效力试验第二次期中分析结果获得附条件批准上市,暂未获得最终分析数据,有效性和安全性尚待进一步确证。
本品基础免疫为 2 剂次,每剂间隔 21-28 天每一次人用剂量为 0.5 ml。
推荐的接种途径为肌内注射,最佳部位为上臂三角肌,注射前须摇匀。
尚未确定本品是否需要进行加强免疫。
本品适用于 18 岁及以上人群预防接种。
境外期临床期中分析保护效力数据显示本品对 18-59 岁人群具有保护作用;临床试验中 60 岁及以上人群所占比例较低(1.57%),后续临床试验中将增加 60 岁及以上人群的比例,进一步获取该人群保护效力直接证据。已有的临床试验数据显示 60 岁及以上人群接种本品后产生一定程度的中和抗体。疾病预防控制相关机构接种时,需结合 60 岁及以上人群健康状况和暴 露风险,以评估接种本品的必要性。
在境内外开展的两项临床试验中评价本品的安全性。第一项为境内随机、双盲、安慰剂平行对照的I/II期临床试验,初步评价本品在18岁及以上人群中的安全性和免疫原性。第二项为国际多中心、随机、双盲、安慰剂平行对照的III期临床试验,评价本品的保护效力、安全性和免疫原性。研究者主动随访每剂接种后0-21/28天的安全性数据,观察不良事件发生情况,同时关注全程接种后12个月内发生的严重不良事件。
1.本品临床试验不良反应发生情况总述
按照国际医学科学组织委员会(CIOMS)推荐的不良反应发生率分类:十分常见(≥10%),常见(1-10%,含1%),偶见(0.1-1%,含0.1%),罕见(0.01-0.1%,含0.01%),十分罕见(<0.01%),汇总本品I/Ⅱ期和III期临床试验研究人群安全性数据进行如下描述:
(1)接种部位不良反应
十分常见:疼痛;
偶见:红晕、肿胀、硬结、皮疹、瘙痒;
罕见:红斑。
(2)全身不良反应
十分常见:头痛;
常见:发热、疲劳/乏力、肌肉痛、关节痛、咳嗽、呼吸困难、恶心、腹泻、皮肤瘙痒;
偶见:头晕、厌食、呕吐、口咽疼痛、吞咽困难、流涕、便秘、超敏反应;
罕见:急性过敏反应、嗜睡、困倦、入睡困难、喷嚏、鼻咽炎、鼻充血、咽干、流行性感冒、感觉减退、肢体疼痛、心悸、腹痛、皮疹、皮肤黏膜异常、痤疮、眼痛、耳部不适、淋巴结病;
十分罕见:寒颤、味觉障碍、味觉丧失、感觉异常、震颤、注意障碍、鼻衄、哮喘、咽喉刺激、扁桃体炎、肢体不适、颈部疼痛、颌骨疼痛、颈部肿块、口腔溃疡、牙疼、食管疾病、胃炎、粪便变色、眼痛、视物模糊、眼刺激、视力减退、耳痛、紧张、高血压、低血压、尿失禁、月经延迟。
(3)不良反应严重程度
本品临床试验中观察到的不良反应严重程度以1级(轻度)为主,3级及以上征集性不良反应发生率为0.44%,未报告与接种本品相关的4级不良反应。临床试验报告的3级接种部位不良反应为疼痛、皮疹、瘙痒;3级全身不良反应为发热、疲劳/乏力、头痛、肌肉痛、关节痛、咳嗽、呼吸困难、呕吐、腹泻、便秘、吞咽困难。
(4)严重不良事件(SAE)
截至2020年10月31日,境外III期临床试验观察到的严重不良事件中,有1例受试者接种本品后出现较为严重的恶心、呕吐等症状,入院经药物治疗后已痊愈,研究者判断与接种本品有关。另有1例受试者接种本品后出现“右上肢无力、口齿不清”症状,就医后当地医院以“炎性脱髓鞘综合症;多发性硬化症(MS);临床孤立综合征(CIS);急性播散性脑脊髓炎(ADEM)待排除”收入院。目前尚无法确定该病例与接种本品的相关性。
2.本品境内外各项临床试验不良反应具体发生情况
(1)境内I/II期临床试验
境内I/II期临床试验共入组1120名18岁及以上受试者,其中420例受试者至少接种1剂本品(I/II期临床试验中剂量组),男性192名(45.71%),女性228人(54.29%)。截至2020年10月13日,已完成全程免疫后至少28天的安全性随访,长期安全性随访尚在进行中。本品不同免疫程序接种后不良反应发生率详见表1。

(2)境外III期临床试验
境外III期临床试验计划入组45000名18岁及以上受试者。截至2020年10月31日期中分析时,14312例受试者至少接种1剂本品。其中,男性12088人(84.46%),女性2224人(15.54%);18~59岁14209人(99.28%),60岁及以上103人(0.72%)。已完成全程免后至少28天的安全性随访,长期安全性随访尚在进行中。III期临床试验18~59岁和60岁及以上人群接种本品的征集性不良反应发生情况详见表2,尚未发现60岁及以上人群安全性风险信号的增加。III期临床试验中观察到的非征集性不良反应发生率为16.03%,3级及以上发生率为0.23%。非征集性不良反应中较征集性不良反应新增严重程度为3级的症状包括:口咽痛(0.01%)、非接种部位皮疹(0.01%)、淋巴结病(0.01%)、超敏反应(0.01%)。


1.对本品所含任何成分(包括辅料)过敏者。
2.既往发生过疫苗严重过敏反应者(如急性过敏反应、血管神经性水肿、呼吸困难等)。
3.患有未控制的癫痫和其他进行性神经系统疾病者,有格林巴利综合征病史者。
4.妊娠期及哺乳期妇女。
5.患急性疾病、严重慢性疾病、慢性疾病的急性发作期和发热者。
6.有惊厥、癫痫、脑病或精神疾病史或家族史。
本品为附条件批准上市。
1.目前暂未获得本品的保护持久性数据, 接种后仍需根据疫情防控需要采取必要的防护措施。
2.目前暂未获得本品对 60 岁及以上人群的保护效力直接证据, 疾病预防控制相关机构接种使用时, 需结合该人群健康状态和暴露风险,评估接种本品的必要性。
3.使用前应检查包装容器、标签、外观有效期是否符合要求,疫苗使用前应充分摇匀,如出现摇不散的凝块、异物、注射器或西林瓶有裂纹或外表面有斑点、污点、擦痕,标签不清或超过有效期时限及外观异常等均不得使用。
4.不得静脉注射;尚无本品采用皮下或皮内注射的安全性和有效性数据。
5.接种本品后应在现场观察至少 30 分钟。接种门诊应备有肾上腺素等急救药物,以备发生严重过敏反应时急救用。
6.患急性疾病、慢性疾病的急性发作期严重慢性疾病、过敏体质和发热者需慎用;必要时经医生评估后延迟接种。
7.糖尿病患者及有惊厥、癫痫、脑病或精神疾病史或家族史者需慎用。
8.血小板减少症及任何凝血功能障碍患者,肌肉注射时可能会引起出血,需慎用。
9.尚未获得本品对免疫功能受损者(例如恶性肿瘤、肾病综合征、艾滋病患者)的安全性和有效性数据,此类人群接种本品应基于个体化考虑。
10.注射过免疫球蛋白者应间隔 1 个月以上再接种本品,以免影响免疫效果。
11.接种本品后出现任何神经系统不良反应者,禁止再次使用。
12.本品尚无 SARS-CoV-2 感染者或既往感染者的保护效力证据。
13.与其它疫苗一样,无法确保本品对所有接种者均产生保护作用。
14.针与针头护帽不得分离。针头护帽表面色泽应均匀,不得有污点、杂质、气泡、裂纹、缺胶或粗糙,护帽内不得有胶丝、胶屑。
育龄期妇女:在临床试验中接种本品后意外妊娠的妇女中收集到的数据非常有限,尚不足以评估接种本品后可能导致发生不良妊娠结局(包括自然流产)的风险。妊娠期或哺乳期女性:目前尚未获得妊娠期及哺乳期妇女使用本品的临床试验数据。60岁及以上人群:目前已在境内I/II期临床试验中获得该人群接种本品的免疫原性与安全性数据,但尚未在境外Ⅲ期临床试验中获得保护效力直接证据。
本节阐述了其他药物对于伏立康唑的影响,伏立康唑对其他药物的影响以及两药间的相互作用。相互作用的第1和第2部分按下列顺序阐述 :禁止合用 ;合用时需要调整剂量并进行密切的临床和/或生物学监测 ;最后是无明显药代动力学相互作用,但可能对临床治疗有益。
北京生物制品研究所有限责任公司
86981597000072;86981597000041;86981597000058;86981597000065
本品关键性Ⅲ期注册临床试验采用多中心随机、双盲、安慰剂平行对照设计,在阿拉伯联合酋长国(阿布扎比、沙迦)、巴林王国等多个国家/地区开展。试验拟入组至少 45000 例 18 岁及以上健康受试者,按照"0,21( + 7)
天"两剂程序随机接种试验疫苗 1(本品)、试验疫苗 2 和安慰剂,以评价本品的保护效力、安全性和免疫原性。主要研究假设为:在 18 岁及以上健康人群中,接种本品 2 剂 14 天后相较于安慰剂的保护效力(VE)的 95% 置信区间(95%Cl)下限大于 30%。本品第二次期中分析结果如下:
1.保护效力试验结果
Ⅲ期临床研究主要终点为接种 2 剂本品或安慰剂 14 天后 COVD-19 的发病率,基于人年发病率的保护率计算方法是疫苗保护效力的主要分析方法。
监测期内所有有效终点病例(120 例)均经过终点判定委员会确认第二次期中分析数据显示,按照 0,21( + 7)天两剂程序接种本品 14 天后预防 COVD-19 的保护效力为 72.51%(双侧 97.59%Cl:54.69,83.32),达到了保护效力研
究假设。接种本品的受试者全程免后平均随访时间为 75.2±27.9 天,中位随访时间为 91 天,结果详见表 3。
境外Ⅲ期临床试验中,可纳入保护效力期中分析的 60 岁及以上人群占 1.57%,目前该人群中尚未收集到终点病例暂无法评价本品对 60 岁及以上人群的保护效力;本次期中分析仅收集到 3 例重症病例,其中安慰剂组 2 例,本品所在组别 1 例,暂无法评价本品对 COVID-19 重症的保护效力。
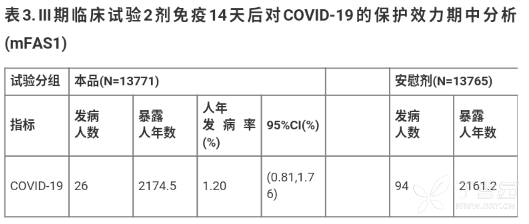
注:(1)基于人年发病率的保护率: Poisson 回归模型以发病人数为因变量,以分组为固定效应,受试者暴露人年数为偏移量,以对数为联结函数。经模型计算各组人年发病率的最小二乘估计及其 95% 可信区间,以及基于人年发病率的保护率的最小二乘估计及其可信区间。(2)基于第二次期
中分析的实际病例数,采用 Lan DeMets O'Brien-Flem-消耗函数方法(PASS 15)计算,单侧检验水准为 0.01205。
2.免疫原性试验结果
Ⅰ/Ⅱ期、Ⅲ期临床研究分别采用噬斑减少中和试验法(PRNT)、微量细胞病变法(CCID)检测免后血清中和抗体几何平均滴度(GMT),结果详见表 4 和表 5。本品基于境外Ⅲ期临床保护效力试验期中分析结果获得附条件批准上 市,Ⅲ期免疫原性亚组免疫持久性数据尚在进一步收集中。现有数据提示,60 岁及以上人群的中和抗体水平低于 18-59 岁人群。
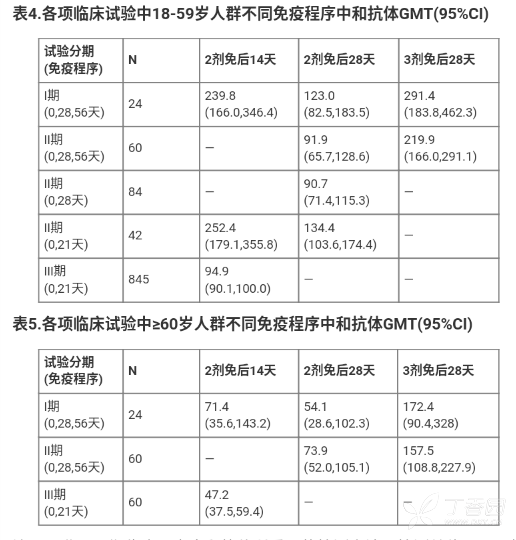
注:Ⅰ/Ⅱ期、Ⅲ期临床研究中和抗体所采用的检测方法、检测单位不同,其检测结果可能存在差异。
在以上试验中获得的安全性数据请参见【不良反应】。
3.交叉中和实验结果
采用境外期临床试验受试者接种本品 2 剂后 28 天的 50 份血清,对目前正在境内流行或国际上具有代表性的 10 株国内外流行的新冠病毒株(2366T、77、76、F13 P4、35T P2、56Y P3、HN7 P3、834 Y、QD01、P701)进行血清交叉中和实验。采用微量细胞病变法进行血清中和抗体检测,数据 显示,50 份血清针对 10 个毒株的中和抗体 GMT 最低为 118.3,最高为 165.5,各毒株间未见明显差异。
注射剂
2.5mL/瓶(5次人用剂量),含灭活新型冠状病毒抗原32.5U;0.5mL/支。每1次人用剂量0.5mL,含灭活新型冠状病毒抗原6.5U;1.0mL/瓶(2次人用剂量),含灭活新型冠状病毒抗原13U。
预灌封注射器组合件(带注射针),1支/盒;中性硼硅玻璃管制注射剂瓶、注射液用覆膜卤化丁基橡胶塞,1瓶/盒、3瓶/盒。
于2~8℃避光保存和运输。避免冻结。
暂定24个月。
国药准字S20217018;国药准字S20200029;国药准字S20200030;国药准字S20217007

微信扫一扫,关注医库最新动态